Getting Started with Nextflow
Overview
Teaching: 30 min
Exercises: 10 minQuestions
What is a workflow and what are workflow management systems?
Why should I use a workflow management system?
What is Nextflow?
What are the main features of Nextflow?
What are the main components of a Nextflow script?
How do I run a Nextflow script?
Objectives
Understand what a workflow management system is.
Understand the benefits of using a workflow management system.
Explain the benefits of using Nextflow as part of your bioinformatics workflow.
Explain the components of a Nextflow script.
Run a Nextflow script.
Workflows
Analysing data involves a sequence of tasks, including gathering, cleaning, and processing data. These sequence of tasks are called a workflow or a pipeline. These workflows typically require executing multiple software packages, sometimes running on different computing environments, such as a desktop or a compute cluster. Traditionally these workflows have been joined together in scripts using general purpose programming languages such as Bash or Python.
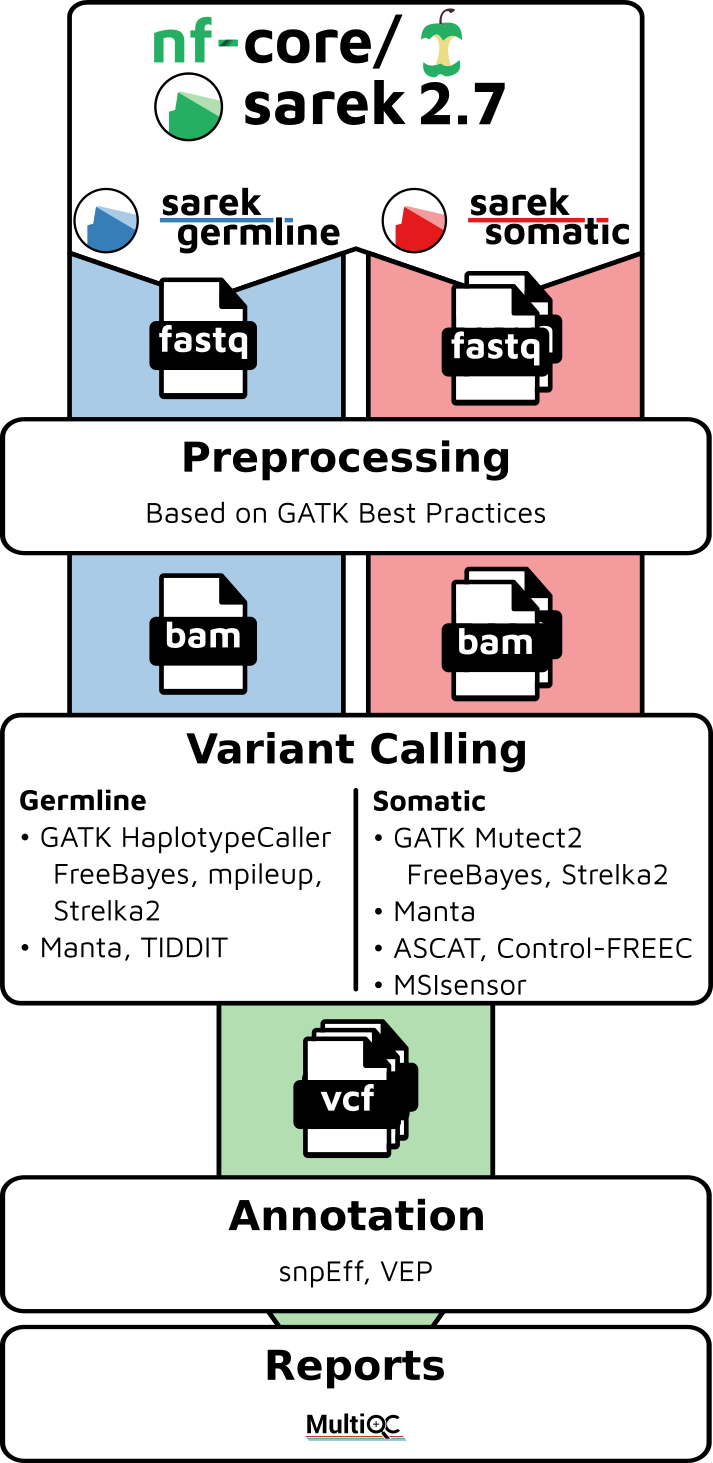
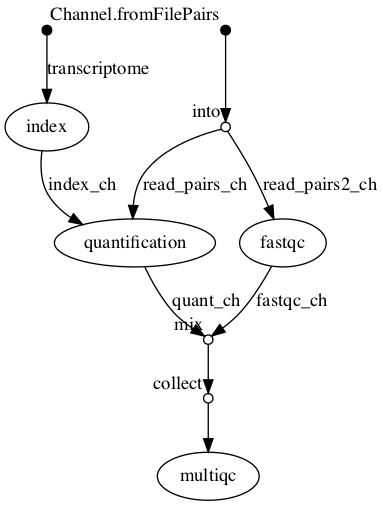
Example bioinformatics variant calling workflow/pipeline diagram from nf-core (https://nf-co.re/sarek) and simple RNA-Seq pipeline in DAG format.
However, as workflows become larger and more complex, the management of the programming logic and software becomes difficult.
Workflow management systems
Workflow Management Systems (WfMS), such as Snakemake, Galaxy, and Nextflow have been developed specifically to manage computational data-analysis workflows in fields such as Bioinformatics, Imaging, Physics, and Chemistry.
WfMS contain multiple features that simplify the development, monitoring, execution and sharing of pipelines.
Key features include;
- Run time management: Management of program execution on the operating system and splitting tasks and data to run at the same time in a process called parallelisation.
- Software management: Use of technology like containers, such as Docker or Singularity, that packages up code and all its dependencies so the application runs reliably from one computing environment to another.
- Portability & Interoperability: Workflows written on one system can be run on another computing infrastructure e.g., local computer, compute cluster, or cloud infrastructure.
- Reproducibility: The use of software management systems and a pipeline specification means that the workflow will produce the same results when re-run, including on different computing platforms.
- Reentrancy: Continuous checkpoints allow workflows to resume from the last successfully executed steps.
Nextflow basic concepts
Nextflow is a workflow management system that combines a runtime environment, software that is designed to run other software, and a programming domain specific language (DSL) that eases the writing of computational pipelines.
Nextflow is built around the idea that Linux is the lingua franca of data science. Nextflow follows Linux’s “small pieces loosely joined” philosophy: in which many simple but powerful command-line and scripting tools, when chained together, facilitate more complex data manipulations.
Nextflow extends this approach, adding the ability to define complex program interactions and an accessible (high-level) parallel computational environment based on the dataflow programming model, whereby processes are connected via their outputs and inputs to other processes, and run as soon as they receive an input.
The diagram below illustrates the differences between a dataflow model and a simple linear program .
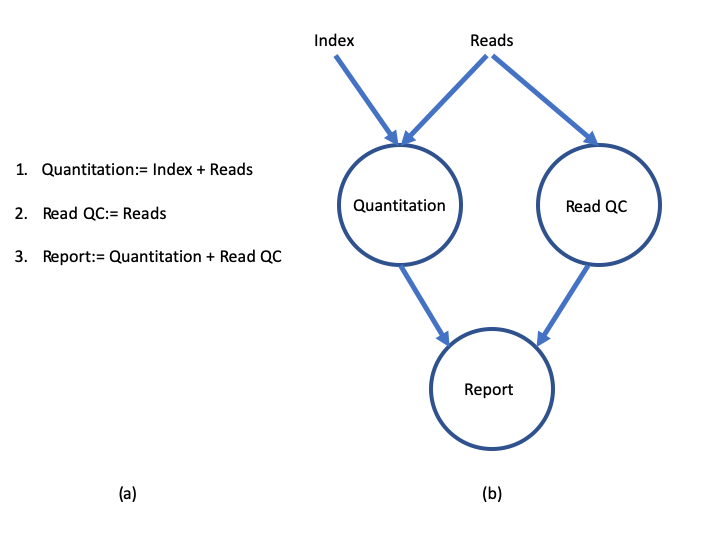
A simple program (a) and its dataflow equivalent (b) https://doi.org/10.1145/1013208.1013209.
In a simple program (a), these statements would be executed sequentially. Thus, the program would execute in three units of time. In the dataflow programming model (b), this program takes only two units of time. This is because the read quantitation and QC steps have no dependencies on each other and therefore can execute simultaneously in parallel.
Nextflow core features
-
Fast prototyping: A simple syntax for writing pipelines that enables you to reuse existing scripts and tools for fast prototyping.
-
Reproducibility: Nextflow supports several container technologies, such as Docker and Singularity, as well as the package manager Conda. This, along with the integration of the GitHub code sharing platform, allows you to write self-contained pipelines, manage versions and to reproduce any former configuration.
-
Portability: Nextflow’s syntax separates the functional logic (the steps of the workflow) from the execution settings (how the workflow is executed). This allows the pipeline to be run on multiple platforms, e.g. local compute vs. a university compute cluster or a cloud service like AWS, without changing the steps of the workflow.
-
Simple parallelism: Nextflow is based on the dataflow programming model which greatly simplifies the splitting of tasks that can be run at the same time (parallelisation).
-
Continuous checkpoints: All the intermediate results produced during the pipeline execution are automatically tracked. This allows you to resume its execution from the last successfully executed step, no matter what the reason was for it stopping.
Scripting language
Nextflow scripts are written using a language intended to simplify the writing of workflows. Languages written for a specific field are called Domain Specific Languages (DSL), e.g., SQL is used to work with databases, and AWK is designed for text processing.
In practical terms the Nextflow scripting language is an extension of the Groovy programming language, which in turn is a super-set of the Java programming language. Groovy simplifies the writing of code and is more approachable than Java. Groovy semantics (syntax, control structures, etc) are documented here.
The approach of having a simple DSL built on top of a more powerful general purpose programming language makes Nextflow very flexible. The Nextflow syntax can handle most workflow use cases with ease, and then Groovy can be used to handle corner cases which may be difficult to implement using the DSL.
DSL2 syntax
Nextflow (version > 20.07.1) provides a revised syntax to the original DSL, known as DSL2. The DSL2 syntax introduces several improvements such as modularity (separating components to provide flexibility and enable reuse), and improved data flow manipulation. This further simplifies the writing of complex data analysis pipelines, and enhances workflow readability, and reusability.
This feature is enabled by the following directive at the beginning a workflow script:
nextflow.enable.dsl=2
Earlier syntax versions
Scripts that contain the directive
nextflow.preview.dsl=2use an early version of the DSL2 syntax, which may include experimental features that have been changed or removed in the formal DSL2 syntax. Scripts without these directives use the first version of the Nextflow syntax which we refer to as DSL1. DSL1 workflows use many of the same concepts presented in this lesson, but some aspects such as the flow of data are written differently. DSL1 workflows are also written in a single script, unlike DSL2 workflows which can be spread across many files. This lesson will focus on the DSL2 syntax as, after the DSL1 to DSL2 transition period is over, it will become the default way of writing Nextflow workflows.
Processes, channels, and workflows
Nextflow workflows have three main parts; processes, channels, and workflows. Processes describe a task to be run. A process script can be written in any scripting language that can be executed by the Linux platform (Bash, Perl, Ruby, Python, etc.). Processes spawn a task for each complete input set. Each task is executed independently, and cannot interact with another task. The only way data can be passed between process tasks is via asynchronous queues, called channels.
Processes define inputs and outputs for a task. Channels are then used to manipulate the flow of data from one process to the next. The interaction between processes, and ultimately the pipeline execution flow itself, is then explicitly defined in a workflow section.
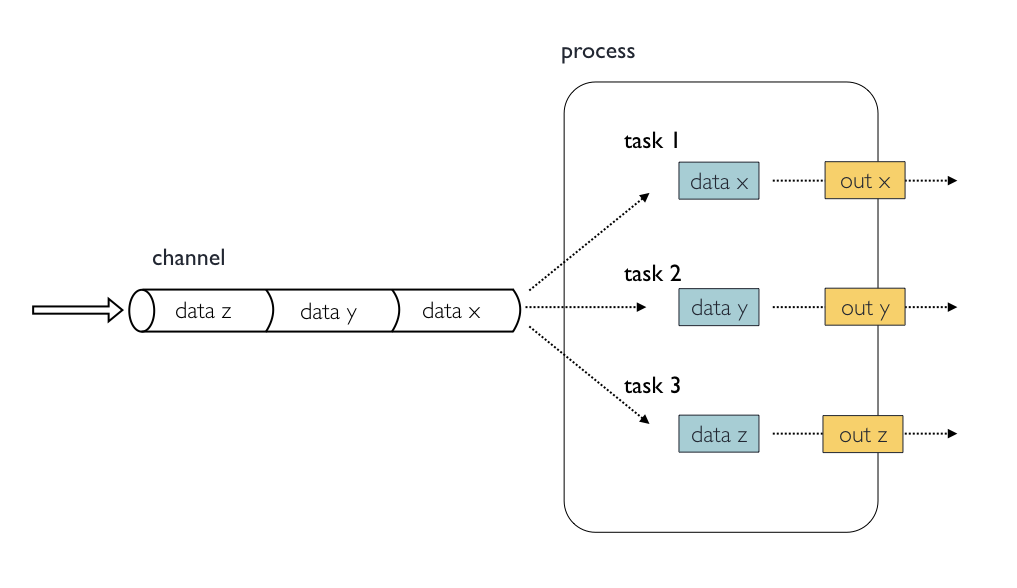
In the following example we have a channel containing three elements, e.g., 3 data files. We have a process that takes the channel as input. Since the channel has three elements, three independent instances (tasks) of that process are run in parallel. Each task generates an output, which is passed to another channel and used as input for the next process.
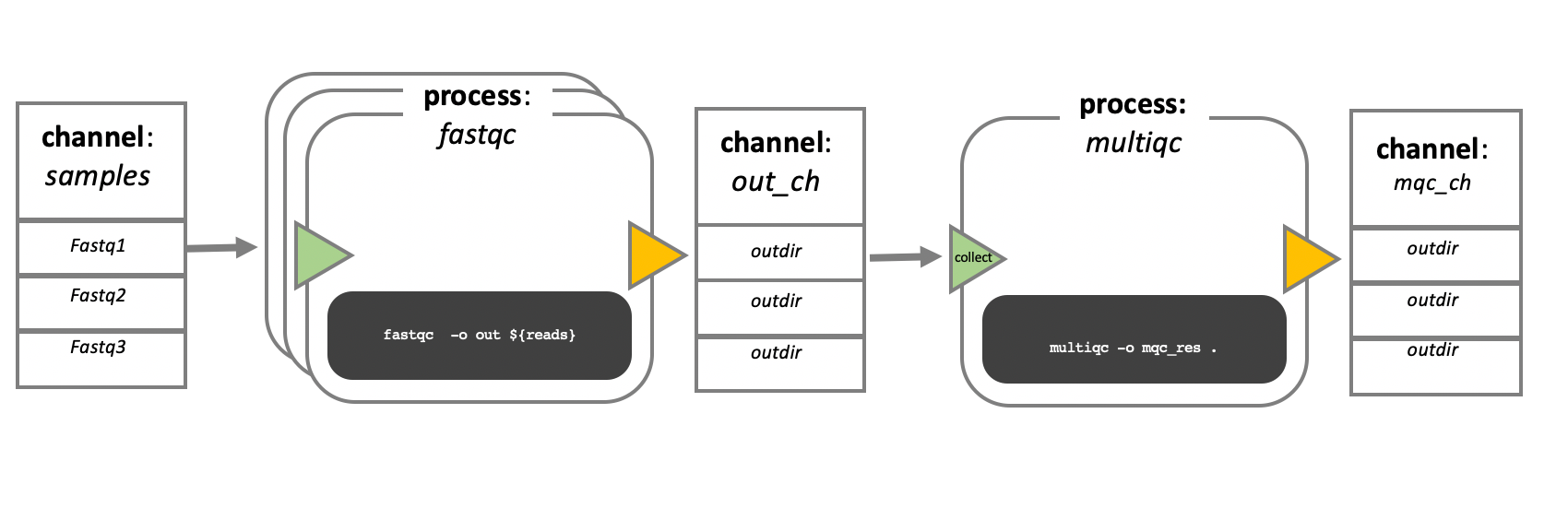
Nextflow process flow diagram
Workflow execution
While a process defines what command or script has to be executed, the executor determines how that script is actually run in the target system.
If not otherwise specified, processes are executed on the local computer. The local executor is very useful for pipeline development, testing, and small scale workflows, but for large scale computational pipelines, a High Performance Cluster (HPC) or Cloud platform is often required.
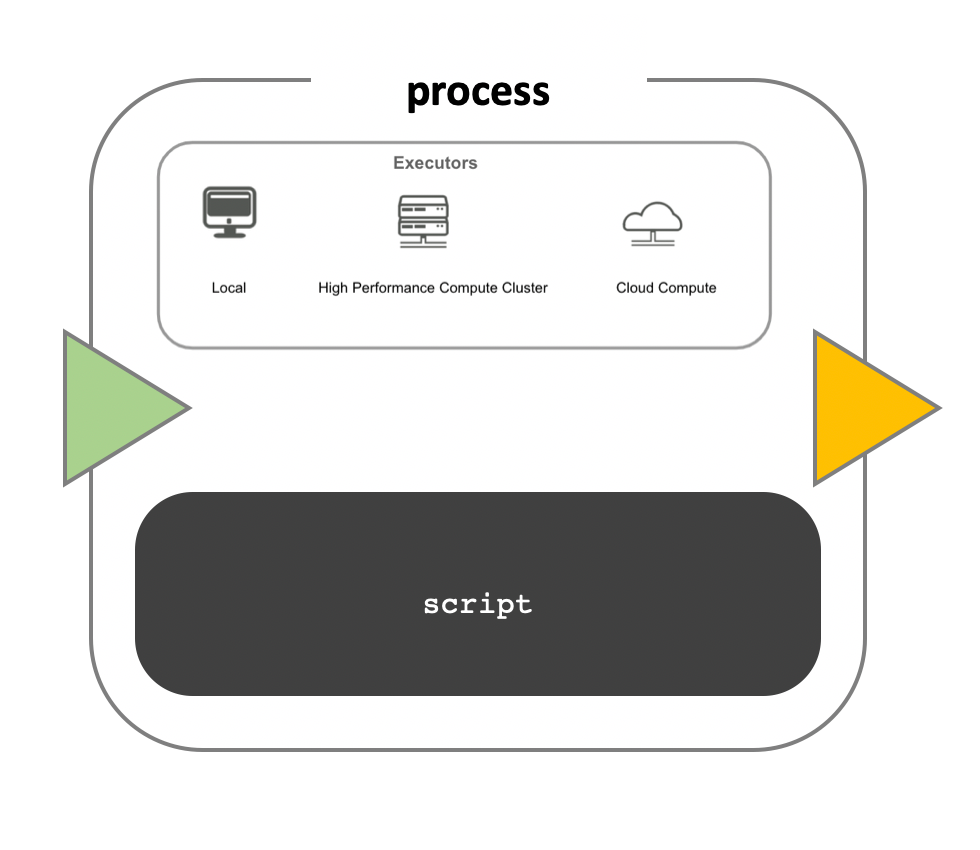
Nextflow Executors
Nextflow provides a separation between the pipeline’s functional logic and the underlying execution platform. This makes it possible to write a pipeline once, and then run it on your computer, compute cluster, or the cloud, without modifying the workflow, by defining the target execution platform in a configuration file.
Nextflow provides out-of-the-box support for major batch schedulers and cloud platforms such as Sun Grid Engine, SLURM job scheduler, AWS Batch service and Kubernetes. A full list can be found here.
Your first script
We are now going to look at a sample Nextflow script that counts the number of lines in a file.
Open the file wc.nf in the script directory with your favourite text editor.
This is a Nextflow script. It contains;
- An optional interpreter directive (“Shebang”) line, specifying the location of the Nextflow interpreter.
nextflow.enable.dsl=2to enable DSL2 syntax.- A multi-line Nextflow comment, written using C style block comments, followed by a single line comment.
- A pipeline parameter
params.inputwhich is given a default value, of the relative path to the location of a compressed fastq file, as a string. - An unnamed
workflowexecution block, which is the default workflow to run. - A Nextflow channel used to read in data to the workflow.
- A call to the process
NUM_LINES. - An operation on the process output, using the channel operator
view(). - A Nextflow
processblock namedNUM_LINES, which defines what the process does. - An
inputdefinition block that assigns the input to the variableread, and declares that it should be interpreted as a filepath. - An
outputdefinition block that uses the Linux/Unix standard output streamstdoutfrom the script block. -
A scriptblock that contains the bash commands ` printf ‘${read}to print the name of the read file, andgunzip -c ${read}wc -l` to count the number of lines in the gzipped read file.
#!/usr/bin/env nextflow
nextflow.enable.dsl=2
/* Comments are uninterpreted text included with the script.
They are useful for describing complex parts of the workflow
or providing useful information such as workflow usage.
Usage:
nextflow run wc.nf --input <input_file>
Multi-line comments start with a slash asterisk /* and finish with an asterisk slash. */
// Single line comments start with a double slash // and finish on the same line
/* Workflow parameters are written as params.<parameter>
and can be initialised using the `=` operator. */
params.input = "data/yeast/reads/ref1_1.fq.gz"
// The default workflow
workflow {
// Input data is received through channels
input_ch = Channel.fromPath(params.input)
/* The script to execute is called by its process name,
and input is provided between brackets. */
NUM_LINES(input_ch)
/* Process output is accessed using the `out` channel.
The channel operator view() is used to print
process output to the terminal. */
NUM_LINES.out.view()
}
/* A Nextflow process block
Process names are written, by convention, in uppercase.
This convention is used to enhance workflow readability. */
process NUM_LINES {
input:
path read
output:
stdout
script:
/* Triple quote syntax """, Triple-single-quoted strings may span multiple lines. The content of the string can cross line boundaries without the need to split the string in several pieces and without concatenation or newline escape characters. */
"""
printf '${read} '
gunzip -c ${read} | wc -l
"""
}
To run a Nextflow script use the command nextflow run <script_name>.
Run a Nextflow script
Run the script by entering the following command in your terminal:
$ nextflow run wc.nfSolution
You should see output similar to the text shown below:
N E X T F L O W ~ version 20.10.0 Launching `wc.nf` [fervent_babbage] - revision: c54a707593 executor > local (1) [21/b259be] process > NUM_LINES (1) [100%] 1 of 1 ✔ ref1_1.fq.gz 58708
- The first line shows the Nextflow version number.
- The second line shows the run name
fervent_babbage(adjective and scientist name) and revision idc54a707593.- The third line tells you the process has been executed locally (
executor > local).- The next line shows the process id
21/b259be, process name, number of cpus, percentage task completion, and how many instances of the process have been run.- The final line is the output of the
viewoperator.
Process identification
The hexadecimal numbers, like 61/1f3ef4, identify the unique process execution. These numbers are also the prefix of the directories where each process is executed. You can inspect the files produced by changing to the directory
$PWD/workand using these numbers to find the process-specific execution path. We will learn how exactly nextflow using work directory to execute processes in the following sections.
Key Points
A workflow is a sequence of tasks that process a set of data.
A workflow management system (WfMS) is a computational platform that provides an infrastructure for the set-up, execution and monitoring of workflows.
Nextflow is a workflow management system that comprises both a runtime environment and a domain specific language (DSL).
Nextflow scripts comprise of channels for controlling inputs and outputs, and processes for defining workflow tasks.
You run a Nextflow script using the
nextflow runcommand.
Nextflow scripting
Overview
Teaching: 30 min
Exercises: 5 minQuestions
What language are Nextflow scripts written in?
How do I store values in a Nextflow script?
How do I write comments in a Nextflow script?
How can I store and retrieve multiple values?
How are strings evaluated in Nextflow?
How can I create simple re-useable code blocks?
Objectives
Understand what language Nextflow scripts are written in.
Define variables in a script.
Create lists of simple values.
Comment Nextflow scripts.
Explain what a list is.
Explain what string interpolation is.
Understand what a closure is.
Nextflow is a Domain Specific Language (DSL) implemented on top of the Groovy programming language, which in turn is a super-set of the Java programming language. This means that Nextflow can run any Groovy and Java code. It is not necessary to learn Groovy to use Nextflow DSL but it can be useful in edge cases where you need more functionality than the DSL provides.
Nextflow console
Nextflow has a console graphical interface. The console is a REPL (read-eval-print loop) environment that allows a user to quickly test part of a script or pieces of Nextflow code in an interactive manner.
It is a handy tool that allows a user to evaluate fragments of Nextflow/Groovy code or fast prototype a complete pipeline script. More information can be found here
We can use the command
nextflow consoleto launch the interactive console to test out out Groovy code.nextflow consoleConsole global scope
It is worth noting that the global script context is maintained across script executions. This means that variables declared in the global script scope are not lost when the script run is complete, and they can be accessed in further executions of the same or another piece of code.
Language Basics
Printing values
To print something is as easy as using the println method (println is a compression of “print line”) and passing the text to print in quotes.
The text is referred to as a string as in a string of characters.
println("Hello, World!")
Hello, World!
Parenthesis for function invocations are optional. Therefore also the following is a valid syntax.
println "Hello, World!"
Hello, World!
Methods
println is a example of a Groovy method. A method is just a block of code which only runs when it is called.
You can pass data, known as parameters, into a method using the method name followed by brackets ().
Methods are used to perform certain actions, and they are also known as functions.
Methods enable us to reuse code: define the code once, and use it many times.
Comments
When we write any code it is useful to document it using comments.
In Nextflow comments use the same syntax as in the C-family programming languages.
This can be confusing for people familiar with the # syntax for commenting in other languages.
// This is a single line comment. Everything after the // is ignored.
/*
Comments can also
span multiple
lines.
*/
Variables
In any programming language, you need to use variables to store different types of information. A variable is a pointer to a space in the computer’s memory that stores the value associated with it.
Variables are assigned using = and can have any value. Groovy is dynamically-typed which means the variable’s data type is based on its value. For example, setting x = 1 means x is an integer number, but if it is later set to x = "hello" then it becomes a String.
Variable scope
When we create a variable using the
x = 1syntax we can access, (scope), it anywhere (globally) in the script. A variable declared in this fashion is sometimes called a public variable.We can also define variables with a data
typee.g.String x="Hello"or with thedefkeyworddef x=1. This effects the accessibility (scope) of the variable. This is called lexical scoping (sometimes known as static scoping) that sets the scope of a variable so that it may only be accessed from within the block of code in which it is defined. A variable declared in this fashion is sometimes called a private variable.
Types of Data
Groovy knows various types of data. four common ones are:
String− These are text literals which are represented in the form of chain of characters enclosed in quotes. For example"Hello World".int− This is used to represent whole numbers. An example is1234.Boolean− This represents a Boolean value which can either betrueorfalse.float- This is used to represent floating point number12.34.
A more complete list can be found here
In the example below, variable my_var has an integer value of 1:
//int − This is used to represent whole numbers.
my_var = 1
To create a variable with a floating point value, we can execute:
//float − This is used to represent floating point numbers.
my_var = 3.1499392
To create a Boolean value we assign the value true or false.
*Note: Do not enclose a Boolean value in quotes or they will be interpreted as a string.
//Boolean − This represents a Boolean value which can either be true or false.
my_var = false
And to create a string, we add single or double quotes around some text.
For example:
//String - These are text literals which are represented in the form of chain of characters
my_var = "chr1"
Multi-line strings
A block of text that span multiple lines can be defined by delimiting it with triple single ''' or double quotes """:
text = """
This is a multi-line string
using triple quotes.
"""
To display the value of a variable to the screen in Groovy, we can use the println method passing the variable name are a parameter.
x = 1
println(x)
1
Slashy strings
Strings can also be defined using the forward slash / character as delimiter. They are known as slashy strings and are useful for defining regular expressions and patterns, as there is no need to escape backslashes e.g \n specifies a new line. As with double quote strings they allow to interpolate variables prefixed with a $ character.
Try the following to see the difference:
x = /ATP1B2\TP53\WRAP53/
println(x)
ATP1B2\TP53\WRAP53
y = 'ATP1B2\TP53\WRAP53'
println(y)
Produces an error as the \ is a special characters that we need to escape.
// use \ to escape
y = 'ATP1B2\\TP53\\WRAP53'
println(y)
ATP1B2\TP53\WRAP53
String interpolation
To use a variable inside a single or multi-line double quoted string "" prefix the variable name with a $ to show it should be interpolated:
chr = "1"
println("processing chromosome $chr")
processing chromosome 1
Note: Variable names inside single quoted strings do not support String interpolation.
chr = "1"
println('processing chromosome $chr')
processing chromosome $chr
Lists
To store multiple values in a variable we can use a List.
A List (also known as array) object can be defined by placing the list items in square brackets and separating items by commas ,:
kmers = [11,21,27,31]
You can access a given item in the list with square-bracket notation []. These positions are numbered starting at 0, so the first element has an index of 0.
kmers = [11,21,27,31]
println(kmers[0])
11
We can use negative numbers as indices in Groovy. They count from the end of the list rather than the front: the index -1 gives us the last element in the list, -2 the second to last, and so on. Because of this, kmers[3] and kmers[-1] point to the same element in our example list.
kmers = [11,21,27,31]
//Lists can also be indexed with negative indexes
println(kmers[3])
println(kmers[-1])
31
31
Lists can also be indexed using a range. A range is a quick way of declaring a list of consecutive sequential numbers.
To define a range use <num1>..<num2> notation.
kmers = [11,21,27,31]
// The first three elements using a range.
println(kmer[0..2])
[11, 21, 27]
String interpolation of list elements
To use an expression like kmer[0..2] inside a double quoted String "" we use the ${expression} syntax, similar to Bash shell scripts.
For example, the expression below without the {}””
kmers = [11,21,27,31]
println("The first three elements in the Lists are. $kmers[0..2]")
would output.
The first three elements in the Lists are. [11, 21, 27, 31][0..2]
We need to enclose the kmers[0..2] expression inside ${} as below to get the correct output.
kmers = [11,21,27,31]
println("The first three elements in the Lists are. ${kmers[0..2]}")
The first three elements in the Lists are. [11, 21, 27]
List methods
Lists have a number of useful methods that can perform operations on their contents. See more here. When using a method on a type of object you need prefix the method with the variable name.
For example, in order to get the length of the list use the list size method:
mylist = [0,1,2]
println(mylist.size())
//inside a string need we need to use the ${} syntax
println("list size is: ${mylist.size()}")
3
list size is: 3
We can use the get method items to retrieve items in a list.
mylist = [0,1,2]
println mylist.get(1)
1
Listed below are a few more common list methods and their output on a simple example.
mylist = [1,2,3]
println mylist
println mylist + [1]
println mylist - [1]
println mylist * 2
println mylist.reverse()
println mylist.collect{ it+3 }
println mylist.unique().size()
println mylist.count(1)
println mylist.min()
println mylist.max()
println mylist.sum()
println mylist.sort()
println mylist.find{it%2 == 0}
println mylist.findAll{it%2 == 0}
[1, 2, 3]
[1, 2, 3, 1]
[2, 3]
[1, 2, 3, 1, 2, 3]
[3, 2, 1]
[4, 5, 6]
3
1
1
3
6
[1, 2, 3]
2
[2]
Create List and retrieve value
Create a list object
listwith the values 1 to 10. Access the fifth element in the list using with square-bracket notation or using thegetmethod and print the resultsSolution
list = [1,2,3,4,5,6,7,8,9,10] //or list = 1..10 println("${list[4]}") //or println("${list.get(4)}")The fifth element is
5. Remember that the array index starts at 0.
Maps
It can difficult to remember the index of a value in a list, so we can use Groovy Maps (also known as associative arrays) that have an arbitrary type of key instead of an integer value. The syntax is very similar to Lists. To specify the key use a colon before the value [key:value]. Multiple values are separated by a comma. Note: the key value is not enclosed in quotes.
roi = [ chromosome : "chr17", start: 7640755, end: 7718054, genes: ['ATP1B2','TP53','WRAP53']]
Maps can be accessed in a conventional square-bracket syntax or as if the key was a property of the map or using the dot notation. Note: When retrieving a value the key value is enclosed in quotes.
//Use of the square brackets.
println(roi['chromosome'])
//Use a dot notation
println(roi.start)
//Use of get method
println(roi.get('genes'))
To add data or to modify a map, the syntax is similar to adding values to list:
//Use of the square brackets
roi['chromosome'] = '17'
//Use a dot notation
roi.chromosome = 'chr17'
//Use of put method
roi.put('genome', 'hg38')
More information about maps can be found in the Groovy API.
Closures
Closures are the swiss army knife of Nextflow/Groovy programming. In a nutshell a closure is a block of code that can be passed as an argument to a function. This can be useful to create a re-usable function.
We can assign a closure to a variable in same way as a value using the =.
square = { it * it }
The curly brackets {} around the expression it * it tells the script interpreter to treat this expression as code. it is an implicit variable that is provided in closures. It’s available when the closure doesn’t have an explicitly declared parameter and represents the value that is passed to the function when it is invoked.
We can pass the function square as an argument to other functions or methods. Some built-in functions take a function like this as an argument. One example is the collect method on lists that iterates through each element of the list transforming it into a new value using the closure:
square = { it * it }
x = [ 1, 2, 3, 4 ]
y = x.collect(square)
println y
[ 1, 4, 9, 16 ]
A closure can also be defined in an anonymous manner, meaning that it is not given a name, and is defined in the place where it needs to be used.
x = [ 1, 2, 3, 4 ]
y = x.collect({ it * it })
println("x is $x")
println("y is $y")
x is [1, 2, 3, 4]
y is [1, 4, 9, 16]
Closure parameters
By default, closures take a single parameter called it. To define a different name use the
` variable ->` syntax.
For example:
square = { num -> num * num }
In the above example the variable num is assigned as the closure input parameter instead of it.
Write a closure
Write a closure to add the prefix
chrto each element of the listx=[1,2,3,4,5,6]Solution
prefix = { "chr${it}"} x = [ 1,2,3,4,5,6 ].collect(prefix) println x[chr1, chr2, chr3, chr4, chr5, chr6]
Multiple map parameters
It’s also possible to define closures with multiple, custom-named parameters using the -> syntax. This separate the custom-named parameters by a comma before the -> operator.
For example:
tp53 = [chromosome: "chr17",start:7661779 ,end:7687538, genome:'GRCh38', gene: "TP53"]
//perform subtraction of end and start coordinates
region_length = {start,end -> end-start }
tp53.length = region_length(tp53.start,tp53.end)
println(tp53)
Would add the region length to the map tp53, calculated as end - start.
[chromosome:chr17, start:7661779, end:7687538, genome:GRCh38, gene:TP53, length:25759]
For another example, the method each() when applied to a map can take a closure with two arguments, to which it passes the key-value pair for each entry in the map object:
//closure with two parameters
printMap = { a, b -> println "$a with value $b" }
//map object
my_map = [ chromosome : "chr17", start : 1, end : 83257441 ]
//each iterates through each element
my_map.each(printMap)
chromosome with value chr17
start with value 1
end with value 83257441
Learn more about closures in the Groovy documentation.
Additional Material
Conditional Execution
If statement
One of the most important features of any programming language is the ability to execute different code under different conditions. The simplest way to do this is to use the if construct.
The if statement uses the syntax common to other programming languages such Java, C, JavaScript, etc.
if( < boolean expression > ) {
// true branch
}
else {
// false branch
}
The else branch is optional. Curly brackets are optional when the branch defines just a single statement.
x = 12
if( x > 10 )
println "$x is greater than 10"
null, empty strings and empty collections are evaluated to false. Therefore a statement like:
list = [1,2,3]
if( list != null && list.size() > 0 ) {
println list
}
else {
println 'The list is empty'
}
Can be written as:
if( list )
println list
else
println 'The list is empty'
In some cases can be useful to replace if statement with a ternary expression, also known as a conditional expression. For example:
println list ? list : 'The list is empty'
The previous statement can be further simplified using the Elvis operator ?: as shown below:
println list ?: 'The list is empty'
For statement
The classical for loop syntax is supported as shown here:
for (int i = 0; i <3; i++) {
println("Hello World $i")
}
Iteration over list objects is also possible using the syntax below:
list = ['a','b','c']
for( String elem : list ) {
println elem
}
Functions
It is possible to define a custom function into a script, as shown here:
int fib(int n) {
return n < 2 ? 1 : fib(n-1) + fib(n-2)
}
println (fib(10)) // prints 89
- A function can take multiple arguments separated by commas.
- The
returnkeyword can be omitted and the function implicitly returns the value of the last evaluated expression. (Not recommended) - Explicit types can be omitted. (Not recommended):
def fact( n ) {
n > 1 ? n * fact(n-1) : 1
}
println (fact(5)) // prints 120
More resources
The complete Groovy language documentation is available at this link.
A great resource to master Apache Groovy syntax is Groovy in Action.
Key Points
Nextflow is a Domain Specific Language (DSL) implemented on top of the Groovy programming language.
To define a variable, assign a value to it e.g.,
a = 1.Comments use the same syntax as in the C-family programming languages:
//or multiline/* */.Multiple values can be stored in lists [value1, value2, value3, …] or maps [chromosome: 1, start :1].
Lists are indexed and sliced with square brackets (e.g., list[0] and list[2..9])
String interpolation (variable interpolation, variable substitution, or variable expansion) is the process of evaluating a string literal containing one or more placeholders, yielding a result in which the placeholders are replaced with their corresponding values.
A closure is an expression (block of code) encased in
{}e.g.{ it * it }.
Workflow parameterization
Overview
Teaching: 20 min
Exercises: 5 minQuestions
How can I change the data a workflow uses?
How can I parameterise a workflow?
How can I add my parameters to a file?
Objectives
Use pipeline parameters to change the input to a workflow.
Add a pipeline parameters to a Nextflow script.
Understand how to create and use a parameter file.
In the first episode we ran the Nextflow script, wc.nf, from the command line and it counted the number of lines in the file
data/yeast/reads/ref1_1.fq.gz. To change the input to script we can make use of pipeline parameters.
Pipeline parameters
The Nextflow wc.nf script defines a pipeline parameter params.input.
Pipeline parameters enable you to change the input to the workflow at
runtime, via the command line or a configuration file, so they are not
hard-coded into the script.
Pipeline parameters are declared in the workflow by prepending the prefix
params, separated by the dot character, to a variable name e.g.,
params.input.
Their value can be specified on the command line by
prefixing the parameter name with a double dash character, e.g., --input.
In the script wc.nf the pipeline parameter params.input was specified with a value of "data/yeast/reads/ref1_1.fq.gz".
To process a different file, e.g. data/yeast/reads/ref2_2.fq.gz, in the wc.nf script we would run:
nextflow run wc.nf --input 'data/yeast/reads/ref2_2.fq.gz'
N E X T F L O W ~ version 21.04.0
Launching `wc.nf` [gigantic_woese] - revision: 8acb5cb9b0
executor > local (1)
[26/3cf986] process > NUM_LINES (1) [100%] 1 of 1 ✔
ref2_2.fq.gz 81720
We can also use wild cards to specify multiple input files (This will be covered in the channels episode).
In the example below we use the * to match any sequence of characters between ref2_ and .fq.gz.
Note: If you use wild card characters on the command line you must enclose the value in quotes.
$ nextflow run wc.nf --input 'data/yeast/reads/ref2_*.fq.gz'
This runs the process NUM_LINES twice, once for each file it matches.
N E X T F L O W ~ version 21.04.0
Launching `wc.nf` [tender_lumiere] - revision: 8acb5cb9b0
executor > local (2)
[cc/b6f793] process > NUM_LINES (1) [100%] 2 of 2 ✔
ref2_2.fq.gz 81720
ref2_1.fq.gz 81720
Change a pipeline’s input using a parameter
Re-run the Nextflow script
wc.nfby changing the pipeline input to all files in the directorydata/yeast/reads/that begin withrefand end with.fq.gz:Solution
$ nextflow run wc.nf --input 'data/yeast/reads/ref*.fq.gz'The string specified on the command line will override the default value of the parameter in the script. The output will look like this:
N E X T F L O W ~ version 20.10.0 Launching `wc.nf` [soggy_miescher] - revision: c54a707593 executor > local (6) [d3/9ca185] process > NUM_LINES (2) [100%] 6 of 6 ✔ ref3_2.fq.gz 52592 ref2_2.fq.gz 81720 ref1_1.fq.gz 58708 ref1_2.fq.gz 58708 ref3_1.fq.gz 52592 ref2_1.fq.gz 81720The pipeline executes the
NUM_LINESprocess 6 times; one process for each file matching the stringdata/yeast/reads/*.fq.gz. Since each process is executed in parallel, there is no guarantee of which output is reported first. When you run this script, you may see the process output in a different order.
Adding a parameter to a script
To add a pipeline parameter to a script prepend the prefix params, separated by a dot character ., to a variable name e.g.,
params.input.
Let’s make a copy of the wc.nf script as wc-params.nf and add a new input parameter.
$ cp wc.nf wc-params.nf
To add a parameter sleep with the default value 2 to wc-params.nf we add the line:
params.sleep = 2
Note: You should always add a sensible default value to the pipeline parameter. We can use this parameter to add another step to our NUM_LINES process.
script:
"""
sleep ${params.sleep}
printf '${read} '
gunzip -c ${read} | wc -l
"""
This step, sleep ${params.sleep}, will add a delay for the amount of time specified in the params.sleep variable,
by default 2 seconds.
To access the value inside the script block we use {variable_name} syntax e.g. ${params.sleep}.
We can now change the sleep parameter from the command line, For Example:
nextflow run wc-params.nf --sleep 10
Add a pipeline parameter
If you haven’t already make a copy of the
wc.nfaswc-params.nf.$ cp wc.nf wc-params.nfAdd the param
sleepwith a default value of 2 below theparams.inputline. Add the linesleep ${params.sleep}in the processNUM_LINESabove the line printf ‘${read}.Run the new script
wc-params.nfchanging the sleep input time.What input file would it run and why?
How would you get it to process all
.fq.gzfiles in thedata/yeast/readsdirectory as well as changing the sleep input to 1 second?Solution
params.sleep=2script: """ sleep ${params.sleep} printf '${read} ' gunzip -c ${read} | wc -l """$ nextflow run wc-params.nf --sleep 1This would use 1 as a value of
sleepparameter instead of default value (which is 2) and run the pipeline. The input file would bedata/yeast/reads/ref1_1.fq.gzas this is the default. To run all input files we could add the param--input 'data/yeast/reads/*.fq.gz'$ nextflow run wc-params.nf --sleep 1 --input 'data/yeast/reads/*.fq.gz'
Parameter File
If we have many parameters to pass to a script it is best to create a parameters file.
Parameters are stored in JSON or YAML format. JSON and YAML are data serialization languages, that are a way of storing data objects and structures, such as the params object in a file.
The -params-file option is used to pass the parameters file to the script.
For example the file wc-params.json contains the parameters sleep and input in JSON format.
{
"sleep": 5,
"input": "data/yeast/reads/etoh60_1*.fq.gz"
}
To run the wc-params.nf script using these parameters we add the option -params-file and pass the file wc-params.json:
$ nextflow run wc-params.nf -params-file wc-params.json
N E X T F L O W ~ version 21.04.0
Launching `wc-params.nf` [nostalgic_northcutt] - revision: 2f86c9ac7e
executor > local (2)
[b4/747eaa] process > NUM_LINES (1) [100%] 2 of 2 ✔
etoh60_1_2.fq.gz 87348
etoh60_1_1.fq.gz 87348
Create and use a Parameter file.
Create a parameter file
params.jsonfor the Nextflow filewc-params.nf, and run the Nextflow script using the created parameter file, specifying:
- sleep as 10
- input as
data/yeast/reads/ref3_1.fq.gzSolution
{ "sleep": 10, "input": "data/yeast/reads/ref3_1.fq.gz" }$ nextflow run wc-params.nf -params-file params.jsonN E X T F L O W ~ version 21.04.0 Launching `wc-params.nf` [small_wiles] - revision: f5ef7b7a01 executor > local (1) [f3/4fa480] process > NUM_LINES (1) [100%] 1 of 1 ✔ ref3_1.fq.gz 52592
Key Points
Pipeline parameters are specified by prepending the prefix
paramsto a variable name, separated by dot character.To specify a pipeline parameter on the command line for a Nextflow run use
--variable_namesyntax.You can add parameters to a JSON or YAML formatted file and pass them to the script using option
-params-file.
Channels
Overview
Teaching: 30 min
Exercises: 10 minQuestions
How do I get data into Nextflow?
How do I handle different types of input, e.g. files and parameters?
How do I create a Nextflow channel?
How can I use pattern matching to select input files?
How do I change the way inputs are handled?
Objectives
Understand how Nextflow manages data using channels.
Understand the different types of Nextflow channels.
Create a value and queue channel using channel factory methods.
Select files as input based on a glob pattern.
Edit channel factory arguments to alter how data is read in.
Channels
Earlier we learnt that channels are the way in which Nextflow sends data around a workflow. Channels connect processes via their inputs and outputs. Channels can store multiple items, such as files (e.g., fastq files) or values. The number of items a channel stores determines how many times a process will run using that channel as input.
Note: When the process runs using one item from the input channel, we will call that run a task.
Why use Channels?
Channels are how Nextflow handles file management, allowing complex tasks to be split up, run in parallel, and reduces ‘admin’ required to get the right inputs to the right parts of the pipeline.
Channels are asynchronous, which means that outputs from a set of processes will not necessarily be produced in the same order as the corresponding inputs went in. However, the first element into a channel queue is the first out of the queue (First in - First out). This allows processes to run as soon as they receive input from a channel. Channels only send data in one direction, from a producer (a process/operator), to a consumer (another process/operator).
Channel types
Nextflow distinguishes between two different kinds of channels: queue channels and value channels.
Queue channel
Queue channels are a type of channel in which data is consumed (used up) to make input for a process/operator. Queue channels can be created in two ways:
- As the outputs of a process.
- Explicitly using channel factory methods such as Channel.of or Channel.fromPath.
DSL1
In Nextflow DSL1 queue channels can only be used once in a workflow, either connecting workflow input to process input, or process output to input for another process. In DSL2 we can use a queue channel multiple times.
Value channels
The second type of Nextflow channel is a value channel. A value channel is bound to a single value. A value channel can be used an unlimited number times since its content is not consumed. This is also useful for processes that need to reuse input from a channel, for example, a reference genome sequence file that is required by multiple steps within a process, or by more than one process.
Queue vs Value Channel.
What type of channel would you use to store the following?
- Multiple values.
- A list with one or more values.
- A single value.
Solution
- A queue channels is used to store multiple values.
- A value channel is used to store a single value, this can be a list with multiple values.
- A value channel is used to store a single value.
Creating Channels using Channel factories
Channel factories are used to explicitly create channels. In programming,
factory methods (functions) are a programming design pattern used
to create different types of objects (in this case, different types
of channels). They are implemented for things that represent more
generalised concepts, such as a Channel.
Channel factories are
called using the Channel.<method> syntax, and return a specific instance
of a Channel.
The value Channel factory
The value factory method is used to create a value channel.
Values are put inside parentheses () to assign them to a channel.
For example:
ch1 = Channel.value( 'GRCh38' )
ch2 = Channel.value( ['chr1', 'chr2', 'chr3', 'chr4', 'chr5'] )
ch3 = Channel.value( ['chr1' : 248956422, 'chr2' : 242193529, 'chr3' : 198295559] )
- Creates a value channel and binds a string to it.
- Creates a value channel and binds a list object to it that will be emitted as a single item.
- Creates a value channel and binds a map object to it that will be emitted as a single item.
The value method can only take 1 argument, however, this can be a single list or map containing several elements.
Reminder:
- A List object can be defined by placing the values in square brackets
[]separated by a comma. - A Map object is similar, but with
key:value pairsseparated by commas.
myList = [1776, -1, 33, 99, 0, 928734928763]
myMap = [ p1 : "start", q2 : "end" ]
Queue channel factory
Queue (consumable) channels can be created using the following channel factory methods.
- Channel.of
- Channel.fromList
- Channel.fromPath
- Channel.fromFilePairs
- Channel.fromSRA
The of Channel factory
When you want to create a channel containing multiple values you can use the channel factory Channel.of. Channel.of allows the creation of a queue channel with the values specified as arguments, separated by a ,.
chromosome_ch = Channel.of( 'chr1', 'chr3', 'chr5', 'chr7' )
chromosome_ch.view()
chr1
chr3
chr5
chr7
The first line in this example creates a variable chromosome_ch. chromosome_ch is a queue channel containing the four values specified as arguments in the of method. The view operator will print one line per item in a list. Therefore the view operator on the second line will print four lines, one for each element in the channel:
You can specify a range of numbers as a single argument using the Groovy range operator ... This creates each value in the range (including the start and end values) as a value in the channel. The Groovy range operator can also produce ranges of dates, letters, or time.
More information on the range operator can be found here.
chromosome_ch = Channel.of(1..22, 'X', 'Y')
chromosome_ch.view()
Arguments passed to the of method can be of varying types e.g., combinations of numbers, strings, or objects. In the above examples we have examples of both string and number data types.
Channel.from
You may see the method
Channel.fromin older nextflow scripts. This performs a similar function but is now deprecated (no longer used), and soChannel.ofshould be used instead.
Create a value and Queue and view Channel contents
- Create a Nextflow script file called
channel.nf.- Create a Value channel
ch_vlcontaining the String'GRCh38'.- Create a Queue channel
ch_qucontaining the values 1 to 4.- Use
.view()operator on the channel objects to view the contents of the channels.- Run the code using
$ nextflow run channel.nfSolution
ch_vl = Channel.value('GRCh38') ch_qu = Channel.of(1,2,3,4) ch_vl.view() ch_qu.view()N E X T F L O W ~ version 21.04.0 Launching `channel.nf` [condescending_dalembert] - revision: c80908867b GRCh38 1 2 3 4
The fromList Channel factory
You can use the Channel.fromList method to create a queue channel from a list object.
aligner_list = ['salmon', 'kallisto']
aligner_ch = Channel.fromList(aligner_list)
aligner_ch.view()
This would produce two lines.
salmon
kallisto
Channel.fromList vs Channel.of
In the above example, the channel has two elements. If you has used the Channel.of(aligner_list) it would have contained only 1 element
[salmon, kallisto]and any operator or process using the channel would run once.
Creating channels from a list
Write a Nextflow script that creates both a
queueandvaluechannel for the listids = ['ERR908507', 'ERR908506', 'ERR908505']Then print the contents of the channels using the
viewoperator. How many lines does the queue and value channel print?Hint: Use the
fromList()andvalue()Channel factory methods.Solution
ids = ['ERR908507', 'ERR908506', 'ERR908505'] queue_ch = Channel.fromList(ids) value_ch = Channel.value(ids) queue_ch.view() value_ch.view()N E X T F L O W ~ version 21.04.0 Launching `channel_fromList.nf` [wise_hodgkin] - revision: 22d76be151 ERR908507 ERR908506 ERR908505 [ERR908507, ERR908506, ERR908505]The queue channel
queue_chwill print three lines.The value channel
value_chwill print one line.
The fromPath Channel factory
The previous channel factory methods dealt with sending general values in a channel. A special channel factory method fromPath is used when wanting to pass files.
The fromPath factory method creates a queue channel containing one or more files matching a file path.
The file path (written as a quoted string) can be the location of a single file or a “glob pattern” that matches multiple files or directories.
The file path can be a relative path (path to the file from the current directory), or an absolute path (path to the file from the system root directory - starts with /).
The script below creates a queue channel with a single file as its content.
read_ch = Channel.fromPath( 'data/yeast/reads/ref1_2.fq.gz' )
read_ch.view()
data/yeast/reads/ref1_2.fq.gz
You can also use glob syntax to specify pattern-matching behaviour for files. A glob pattern is specified as a string and is matched against directory or file names.
- An asterisk,
*, matches any number of characters (including none). - Two asterisks,
**, works like * but will also search sub directories. This syntax is generally used for matching complete paths. - Braces
{}specify a collection of subpatterns. For example:{bam,bai}matches “bam” or “bai”
For example the script below uses the *.fq.gz pattern to create a queue channel that contains as many items as there are files with .fq.gz extension in the data/yeast/reads folder.
read_ch = Channel.fromPath( 'data/yeast/reads/*.fq.gz' )
read_ch.view()
data/yeast/reads/ref1_2.fq.gz
data/yeast/reads/etoh60_3_2.fq.gz
data/yeast/reads/temp33_1_2.fq.gz
data/yeast/reads/temp33_2_1.fq.gz
data/yeast/reads/ref2_1.fq.gz
data/yeast/reads/temp33_3_1.fq.gz
[..truncated..]
Note The pattern must contain at least a star wildcard character.
You can change the behaviour of Channel.fromPath method by changing its options. A list of .fromPath options is shown below.
Available fromPath options:
| Name | Description |
|---|---|
| glob | When true, the characters *, ?, [] and {} are interpreted as glob wildcards, otherwise they are treated as literal characters (default: true) |
| type | The type of file paths matched by the string, either file, dir or any (default: file) |
| hidden | When true, hidden files are included in the resulting paths (default: false) |
| maxDepth | Maximum number of directory levels to visit (default: no limit) |
| followLinks | When true, symbolic links are followed during directory tree traversal, otherwise they are managed as files (default: true) |
| relative | When true returned paths are relative to the top-most common directory (default: false) |
| checkIfExists | When true throws an exception if the specified path does not exist in the file system (default: false) |
We can change the default options for the fromPath method to give an error if the file doesn’t exist using the checkIfExists parameter. In Nextflow, method parameters are separated by a , and parameter values specified with a colon :.
If we execute a Nextflow script with the contents below, it will run and not produce an output. This is likely not what we want.
read_ch = Channel.fromPath( 'data/chicken/reads/*.fq.gz' )
read_ch.view()
Add the argument checkIfExists with the value true.
read_ch = Channel.fromPath( 'data/chicken/reads/*.fq.gz', checkIfExists: true )
read_ch.view()
This will give an error as there is no data/chicken directory.
N E X T F L O W ~ version 20.10.0
Launching `hello.nf` [intergalactic_mcclintock] - revision: d2c138894b
No files match pattern `*.fq.gz` at path: data/chicken/reads/
Using Channel.fromPath
- Create a Nextflow script
channel_fromPath.nf- Use the
Channel.fromPathmethod to create a channel containing all files in thedata/yeast/directory, including the subdirectories.- Add the parameter to include any hidden files.
- Then print all file names using the
viewoperator.Hint: You need two asterisks, i.e.
**, to search subdirectories.Solution
all_files_ch = Channel.fromPath('data/yeast/**', hidden: true) all_files_ch.view()N E X T F L O W ~ version 21.04.0 Launching `nf-training/scripts/channels/channel_fromPath.nf` [reverent_mclean] - revision: cf02269bcb data/yeast/samples.csv data/yeast/bams/ref1.bam.bai data/yeast/bams/ref3.bam data/yeast/bams/etoh60_3. [..truncated..]
The fromFilePairs Channel factory
We have seen how to process files individually using fromPath. In Bioinformatics we often want to process files in pairs or larger groups, such as read pairs in sequencing.
For example is the data/yeast/reads directory we have nine groups of read pairs.
| Sample group | read1 | read2 |
|---|---|---|
| ref1 | data/yeast/reads/ref1_1.fq.gz | data/yeast/reads/ref1_2.fq.gz |
| ref2 | data/yeast/reads/ref2_1.fq.gz | data/yeast/reads/ref2_2.fq.gz |
| ref3 | data/yeast/reads/ref3_1.fq.gz | data/yeast/reads/ref3_2.fq.gz |
| temp33_1 | data/yeast/reads/temp33_1_1.fq.gz | data/yeast/reads/temp33_1_2.fq.gz |
| temp33_2 | data/yeast/reads/temp33_2_1.fq.gz | data/yeast/reads/temp33_2_2.fq.gz |
| temp33_3 | data/yeast/reads/temp33_3_1.fq.gz | data/yeast/reads/temp33_3_2.fq.gz |
| etoh60_1 | data/yeast/reads/etoh60_1_1.fq.gz | data/yeast/reads/etoh60_1_2.fq.gz |
| etoh60_2 | data/yeast/reads/etoh60_2_1.fq.gz | data/yeast/reads/etoh60_2_2.fq.gz |
| etoh60_3 | data/yeast/reads/etoh60_3_1.fq.gz | data/yeast/reads/etoh60_3_2.fq.gz |
Nextflow provides a convenient Channel factory method for this common bioinformatics use case. The fromFilePairs method creates a queue channel containing a tuple for every set of files matching a specific glob pattern (e.g., /path/to/*_{1,2}.fq.gz).
A tuple is a grouping of data, represented as a Groovy List.
- The first element of the tuple emitted from
fromFilePairsis a string based on the shared part of the filenames (i.e., the*part of the glob pattern). - The second element is the list of files matching the remaining part of the glob pattern (i.e., the
<string>_{1,2}.fq.gzpattern). This will include any files ending_1.fq.gzor_2.fq.gz.
read_pair_ch = Channel.fromFilePairs('data/yeast/reads/*_{1,2}.fq.gz')
read_pair_ch.view()
[etoh60_3, [data/yeast/reads/etoh60_3_1.fq.gz, data/yeast/reads/etoh60_3_2.fq.gz]]
[temp33_1, [data/yeast/reads/temp33_1_1.fq.gz, data/yeast/reads/temp33_1_2.fq.gz]]
[ref1, [data/yeast/reads/ref1_1.fq.gz, data/yeast/reads/ref1_2.fq.gz]]
[ref2, [data/yeast/reads/ref2_1.fq.gz, data/yeast/reads/ref2_2.fq.gz]]
[temp33_2, [data/yeast/reads/temp33_2_1.fq.gz, data/yeast/reads/temp33_2_2.fq.gz]]
[ref3, [data/yeast/reads/ref3_1.fq.gz, data/yeast/reads/ref3_2.fq.gz]]
[temp33_3, [data/yeast/reads/temp33_3_1.fq.gz, data/yeast/reads/temp33_3_2.fq.gz]]
[etoh60_1, [data/yeast/reads/etoh60_1_1.fq.gz, data/yeast/reads/etoh60_1_2.fq.gz]]
[etoh60_2, [data/yeast/reads/etoh60_2_1.fq.gz, data/yeast/reads/etoh60_2_2.fq.gz]]
This will produce a queue channel, read_pair_ch , containing nine elements.
Each element is a tuple that has;
- string value (the file prefix matched, e.g
temp33_1) - and a list with the two files e,g.
[data/yeast/reads/temp33_1_1.fq.gz, data/yeast/reads/temp33_1_2.fq.gz].
The asterisk character *, matches any number of characters (including none), and the {} braces specify a collection of subpatterns. Therefore the *_{1,2}.fq.gz pattern matches any file name ending in _1.fq.gz or _2.fq.gz .
What if you want to capture more than a pair?
If you want to capture more than two files for a pattern you will need to change the default size argument (the default value is 2) to the number of expected matching files.
For example in the directory data/yeast/reads there are six files with the prefix ref.
If we want to group (create a tuple) for all of these files we could write;
read_group_ch = Channel.fromFilePairs('data/yeast/reads/ref{1,2,3}*',size:6)
read_group_ch.view()
The code above will create a queue channel containing one element. The element is a tuple of which contains a string value, that is the pattern ref, and a list of six files matching the pattern.
[ref, [data/yeast/reads/ref1_1.fq.gz, data/yeast/reads/ref1_2.fq.gz, data/yeast/reads/ref2_1.fq.gz, data/yeast/reads/ref2_2.fq.gz, data/yeast/reads/ref3_1.fq.gz, data/yeast/reads/ref3_2.fq.gz]]
See more information about the channel factory fromFilePairs here
More complex patterns
If you need to match more complex patterns you should create a sample sheet specifying the files and create a channel from that. This will be covered in the operator episode.
Create a channel containing groups of files
- Create a Nextflow script file
channel_fromFilePairs.nf.- Use the
fromFilePairsmethod to create a channel containing three tuples. Each tuple will contain the pairs of fastq reads for the three temp33 samples in thedata/yeast/readsdirectorySolution
pairs_ch = Channel.fromFilePairs('data/yeast/reads/temp33*_{1,2}.fq.gz') pairs_ch.view()N E X T F L O W ~ version 21.04.0 Launching `channels.nf` [stupefied_lumiere] - revision: a3741edde2 [temp33_1, [data/yeast/reads/temp33_1_1.fq.gz, data/yeast/reads/temp33_1_2.fq.gz]] [temp33_3, [data/yeast/reads/temp33_3_1.fq.gz, data/yeast/reads/temp33_3_2.fq.gz]] [temp33_2, [data/yeast/reads/temp33_2_1.fq.gz, data/yeast/reads/temp33_2_2.fq.gz]]
The fromSRA Channel factory
Another useful factory method is fromSRA. The fromSRA method makes it possible to query the NCBI SRA archive and returns a queue channel emitting the FASTQ files matching the specified selection criteria.
The queries can be project IDs or accession numbers supported by the NCBI ESearch API.
If you want to use this functionality, you will need an NCBI API KEY, and to set the environment variable NCBI_API_KEY to its value.
sra_ch =Channel.fromSRA('SRP043510')
sra_ch.view()
This will print a tuple for every fastq file associated with that SRA project accession.
[SRR1448794, ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR144/004/SRR1448794/SRR1448794.fastq.gz]
[SRR1448795, ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR144/005/SRR1448795/SRR1448795.fastq.gz]
[SRR1448792, ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR144/002/SRR1448792/SRR1448792.fastq.gz]
[SRR1448793, ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR144/003/SRR1448793/SRR1448793.fastq.gz]
[SRR1910483, ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR191/003/SRR1910483/SRR1910483.fastq.gz]
[SRR1910482, ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR191/002/SRR1910482/SRR1910482.fastq.gz]
(remaining omitted)
Multiple accession IDs can be specified using a list object:
ids = ['ERR908507', 'ERR908506', 'ERR908505']
sra_ch = Channel.fromSRA(ids)
sra_ch.view()
[ERR908507, [ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR908/ERR908507/ERR908507_1.fastq.gz, ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR908/ERR908507/ERR908507_2.fastq.gz]]
[ERR908506, [ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR908/ERR908506/ERR908506_1.fastq.gz, ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR908/ERR908506/ERR908506_2.fastq.gz]]
[ERR908505, [ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR908/ERR908505/ERR908505_1.fastq.gz, ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR908/ERR908505/ERR908505_2.fastq.gz]]
Read pairs from SRA
Read pairs are implicitly managed, and are returned as a list of files.
Key Points
Channels must be used to import data into Nextflow.
Nextflow has two different kinds of channels: queue channels and value channels.
Data in value channels can be used multiple times in workflow.
Data in queue channels are consumed when they are used by a process or an operator.
Channel factory methods, such as
Channel.of, are used to create channels.Channel factory methods have optional parameters e.g.,
checkIfExists, that can be used to alter the creation and behaviour of a channel.
Processes
Overview
Teaching: 30 min
Exercises: 15 minQuestions
How do I run tasks/processes in Nextflow?
How do I get data, files and values, into a processes?
Objectives
Understand how Nextflow uses processes to execute tasks.
Create a Nextflow process.
Define inputs to a process.
Processes
We now know how to create and use Channels to send data around a workflow. We will now see how to run tasks within a workflow using processes.
A process is the way Nextflow executes commands you would run on the command line or custom scripts.
A process can be thought of as a particular step in a workflow, e.g. an alignment step in RNA-seq analysis. Processes are independent of each other (don’t require any another process to execute) and can not communicate/write to each other. Data is passed between processes via input and output Channels.
For example, below is the command you would run to count the number of sequence records in a FASTA format file such as the yeast transcriptome:
FASTA format
FASTA format is a text-based format for representing either nucleotide sequences or peptide sequences. A sequence in FASTA format begins with a single-line description, followed by lines of sequence data. The description line is distinguished from the sequence data by a greater-than (“>”) symbol in the first column.
>YBR024W_mRNA cdna chromosome:R64-1-1:II:289445:290350:1 gene:YBR024W gene_biotype:protein_coding transcript_biotype:protein_coding gene_symbol:SCO2 description:Protein anchored to mitochondrial inner membrane; may have a redundant function with Sco1p in delivery of copper to cytochrome c oxidase; interacts with Cox2p; SCO2 has a paralog, SCO1, that arose from the whole genome duplication [Source:SGD;Acc:S000000228] ATGTTGAATAGTTCAAGAAAATATGCTTGTCGTTCCCTATTCAGACAAGCGAACGTCTCA ATAAAAGGACTCTTTTATAATGGAGGCGCATATCGAAGAGGGTTTTCAACGGGATGTTGT
$ zgrep -c '^>' data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz
6612
Now we will show how to convert this into a simple Nextflow process.
Process definition
The process definition starts with keyword process, followed by process name, in this case NUMSEQ, and finally the process body delimited by curly brackets {}. The process body must contain a string which represents the command or, more generally, a script that is executed by it.
process NUMSEQ {
script:
"zgrep -c '^>' ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
}
This process would run once.
Implicit variables
We use the Nextflow implicit variable
${projectDir}to specify the directory where the main script is located. This is important as Nextflow scripts are executed in a separate working directory. A full list of implicit variables can be found here
To add the process to a workflow add a workflow block, and call the process like a function. We will learn more about the workflow block in the workflow episode.
Note: As we are using DSL2 we need to include nextflow.enable.dsl=2 in the script.
//process_01.nf
nextflow.enable.dsl=2
process NUMSEQ {
script:
"zgrep -c '^>' ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
}
workflow {
//process is called like a function in the workflow block
NUMSEQ()
}
We can now run the process:
$ nextflow run process_01.nf -process.echo
N E X T F L O W ~ version 21.10.6
Launching `process_01.nf` [modest_pike] - revision: 3eaa812b17
executor > local (1)
[cd/eab1fd] process > NUMSEQ [100%] 1 of 1 ✔
6612
A Simple Process
Create a Nextflow script
simple_process.nfthat has one processCOUNT_BASESthat runs the command.zgrep -v '^>' ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz|tr -d '\n'|wc -mSolution
nextflow.enable.dsl=2 process COUNT_BASES { script: """ zgrep -v '^>' ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz|tr -d '\n'|wc -m """ } workflow { COUNT_BASES() }Note We need to add the Nextflow run option
-process.echoto print the output to the terminal.$ nextflow run simple_process.nf -process.echoN E X T F L O W ~ version 21.04.0 Launching `simple_process.nf`` [prickly_gilbert] - revision: 471a79c65c executor > local (1) [56/5e6001] process > COUNT_BASES [100%] 1 of 1 ✔ 8772368
Definition blocks
The previous example was a simple process with no defined inputs and outputs that ran only once. To control inputs, outputs and how a command is executed a process may contain five definition blocks:
- directives - 0, 1, or more: allow the definition of optional settings that affect the execution of the current process e.g. the number of cpus a task uses and the amount of memory allocated.
- inputs - 0, 1, or more: Define the input dependencies, usually channels, which determines the number of times a process is executed.
- outputs - 0, 1, or more: Defines the output channels used by the process to send results/data produced by the process.
- when clause - optional: Allows you to define a condition that must be verified in order to execute the process.
- script block - required: A statement within quotes that defines the commands that are executed by the process to carry out its task.
The syntax is defined as follows:
process < NAME > {
[ directives ]
input:
< process inputs >
output:
< process outputs >
when:
< condition >
[script|shell|exec]:
< user script to be executed >
}
Script
At minimum a process block must contain a script block.
The script block is a String “statement” that defines the command that is executed by the process to carry out its task. These are normally the commands you would run on a terminal.
A process contains only one script block, and it must be the last statement when the process contains input and output declarations.
The script block can be a simple one line string in quotes e.g.
nextflow.enable.dsl=2
process NUMSEQ {
script:
"zgrep -c '^>' ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
}
workflow {
NUMSEQ()
}
Or, for commands that span multiple lines you can encase the command in triple quotes """.
For example:
//process_multi_line.nf
nextflow.enable.dsl=2
process NUMSEQ_CHR {
script:
"""
zgrep '^>' ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz > ids.txt
zgrep -c '>YA' ids.txt
"""
}
workflow {
NUMSEQ_CHR()
}
$ nextflow run process_multi_line.nf -process.echo
N E X T F L O W ~ version 21.10.6
Launching `process_multi_line.nf` [focused_jang] - revision: e32caf0dcb
executor > local (1)
[00/14ce67] process > CHR_COUNT (1) [100%] 1 of 1 ✔
Number of sequences for chromosome A:118
By default the process command is interpreted as a Bash script. However, any other scripting language can be used just simply starting the script with the corresponding Shebang declaration. For example:
//process_python.nf
nextflow.enable.dsl=2
process PYSTUFF {
script:
"""
#!/usr/bin/env python
import gzip
reads = 0
bases = 0
with gzip.open('${projectDir}/data/yeast/reads/ref1_1.fq.gz', 'rb') as read:
for id in read:
seq = next(read)
reads += 1
bases += len(seq.strip())
next(read)
next(read)
print("reads", reads)
print("bases", bases)
"""
}
workflow {
PYSTUFF()
}
//process_rscript.nf
nextflow.enable.dsl=2
process RSTUFF {
script:
"""
#!/usr/bin/env Rscript
library("ShortRead")
countFastq(dirPath="data/yeast/reads/ref1_1.fq.gz")
"""
}
workflow {
RSTUFF()
}
This allows the use of a different programming languages which may better fit a particular job. However, for large chunks of code it is suggested to save them into separate files and invoke them from the process script.
nextflow.enable.dsl=2
process PYSTUFF {
script:
"""
myscript.py
"""
}
workflow {
PYSTUFF()
}
Associated scripts
Scripts such as the one in the example above,
myscript.py, can be stored in abinfolder at the same directory level as the Nextflow workflow script that invokes them, and given execute permission. Nextflow will automatically add this folder to thePATHenvironment variable. To invoke the script in a Nextflow process, simply use its filename on its own rather than invoking the interpreter e.g.myscript.pyinstead ofpython myscript.py.
Script parameters
The command in the script block can be defined dynamically using Nextflow variables e.g. ${projectDir}.
To reference a variable in the script block you can use the $ in front of the Nextflow variable name, and additionally you can add {} around the variable name e.g. ${projectDir}.
Variable substitutions
Similar to bash scripting Nextflow uses the “$” character to introduce variable substitutions. The variable name to be expanded may be enclosed in braces
{variable_name}, which are optional but serve to protect the variable to be expanded from characters immediately following it which could be interpreted as part of the name. It is a good rule of thumb to always use the{}syntax.
In the example below the variable chr is set to the value A at the top of the Nextflow script.
The variable is referenced using the $chr syntax within the multi-line string statement in the script block.
A Nextflow variable can be used multiple times in the script block.
//process_script.nf
nextflow.enable.dsl=2
chr = "A"
process CHR_COUNT {
script:
"""
printf "Number of sequences for chromosome ${chr} :"
zgrep -c '>Y'${chr} ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz
"""
}
workflow {
CHR_COUNT()
}
In most cases we do not want to hard code parameter values. We saw in the parameter episode the use of a special Nextflow variable params that can be used to assign values from the command line. You would do this by adding a key name to the params variable and specifying a value, like params.keyname = value
In the example below we define the variable params.chr with a default value of A in the Nextflow script.
//process_script_params.nf
nextflow.enable.dsl=2
params.chr = "A"
process CHR_COUNT {
script:
"""
printf 'Number of sequences for chromosome '${params.chr}':'
zgrep -c '^>Y'${params.chr} ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz
"""
}
workflow {
CHR_COUNT()
}
Remember, we can change the default value of chr to a different value such as B, by running the Nextflow script using the command below. Note: parameters to the workflow have two hyphens --.
nextflow run process_script_params.nf --chr B
Script parameters
For the Nextflow script below.
//process_exercise_script_params.nf nextflow.enable.dsl=2 process COUNT_BASES { script: """ zgrep -v '^>' ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz|grep -o A|wc -l """ } workflow { COUNT_BASES() }Add a parameter params.base to the script and uses the variable ${param.base} insides the script. Run the pipeline using a base value of
Cusing the--basecommand line option.$ nextflow run process_script_params.nf --base <some value> -process.echoNote: The Nextflow option
-process.echowill print the process’ stdout to the terminal.Solution
//process_exercise_script_params.nf nextflow.enable.dsl=2 params.base='A' process COUNT_BASES { script: """ zgrep -v '^>' ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz|grep -o ${params.base}|wc -l """ } workflow { COUNT_BASES() }nextflow run process_script_params.nf --base C -process.echoN E X T F L O W ~ version 21.04.0 Launching `process_script_params.nf ` [nostalgic_jones] - revision: 9feb8de4fe executor > local (1) [92/cdc9de] process > COUNT_BASES [100%] 1 of 1 ✔ 1677188
Bash variables
Nextflow uses the same Bash syntax for variable substitutions, $variable, in strings.
However, Bash variables need to be escaped using \ character in front of \$variable name.
In the example below we will set a bash variable NUMIDS then echo the value of NUMIDS in our script block.
nextflow.enable.dsl=2
process NUM_IDS {
script:
"""
#set bash variable NUMIDS
NUMIDS=`zgrep -c '^>' $params.transcriptome`
echo 'Number of sequences'
printf "%'d\n" \$NUMIDS
"""
}
params.transcriptome = "${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
workflow {
NUM_IDS()
}
Shell
Another alternative is to use a shell block definition instead of script.
When using the shell statement Bash variables are referenced in the normal way $my_bash_variable;
However, the shell statement uses a different syntax for Nextflow variable substitutions: !{nextflow_variable}, which is needed to use both Nextflow and Bash variables in the same script.
For example in the script below that uses the shell statement
we reference the Nextflow variables as !{projectDir} , and the Bash variable as ${NUMCHAR} and ${NUMLINES}.
//process_shell.nf
nextflow.enable.dsl=2
process NUM_IDS {
shell:
//Shell script definition requires the use of single-quote ' delimited strings
'''
#set bash variable NUMIDS
NUMIDS=`zgrep -c '^>' !{params.transcriptome}`
echo 'Number of sequences'
printf $NUMIDS
'''
}
params.transcriptome = "${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
workflow {
NUM_IDS()
}
Conditional script execution
Sometimes you want to change how a process is run depending on some condition. In Nextflow scripts we can use conditional statements such as the if statement or any other expression evaluating to boolean value true or false.
If statement
The if statement uses the same syntax common to other programming languages such Java, C, JavaScript, etc.
if( < boolean expression > ) {
// true branch
}
else if ( < boolean expression > ) {
// true branch
}
else {
// false branch
}
For example, the Nextflow script below will use the if statement to change what the COUNT process counts depending on the Nextflow variable params.method.
//process_conditional.nf
nextflow.enable.dsl=2
params.method = 'ids'
params.transcriptome = "$projectDir/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
process COUNT {
script:
if( params.method == 'ids' ) {
"""
echo Number of sequences in transciptome
zgrep -c "^>" $params.transcriptome
"""
}
else if( params.method == 'bases' ) {
"""
echo Number of bases in transciptome
zgrep -v "^>" $params.transcriptome|grep -o "."|wc -l
"""
}
else {
"""
echo Unknown method $params.method
"""
}
}
workflow {
COUNT()
}
nextflow run process_conditional.nf -process.echo --method ids
N E X T F L O W ~ version 21.04.0
Launching `juggle_processes.nf` [cheeky_shirley] - revision: 588f20ae5a
[01/60b08d] process > COUNT [100%] 1 of 1 ✔
Number of sequences in transciptome
6612
Inputs
Processes are isolated from each other but can communicate by sending values and files via Nextflow channels from input and into output blocks.
The input block defines which channels the process is expecting to receive input from.
The number of elements in input channels determines the process dependencies and the number of times a process executes.
You can only define one input block at a time and it must contain one or more input declarations.
The input block follows the syntax shown below:
input:
<input qualifier> <input name>
The input qualifier declares the type of data to be received.
Input qualifiers
val: Lets you access the received input value by its name as a variable in the process script.env: Lets you use the input value to set an environment variable named as the specified input name.path: Lets you handle the received value as a file, staging the file properly in the execution context.stdin: Lets you forward the received value to the process stdin special file.tuple: Lets you handle a group of input values having one of the above qualifiers.each: Lets you execute the process for each entry in the input collection. A complete list of inputs can be found here.
Input values
The val qualifier allows you to receive value data as input. It can be accessed in the process script by using the specified input name, as shown in the following example:
//process_input_value.nf
nextflow.enable.dsl=2
process PRINTCHR {
input:
val chr
script:
"""
echo processing chromosome $chr
"""
}
chr_ch = Channel.of( 'A' .. 'P' )
workflow {
PRINTCHR(chr_ch)
}
$ nextflow run process_input_value.nf -process.echo
N E X T F L O W ~ version 21.04.0
Launching `process_input_value.nf` [wise_kalman] - revision: 7f90e1bfc5
executor > local (24)
[b1/88df3f] process > PRINTCHR (16) [100%] 24 of 24 ✔
processing chromosome C
processing chromosome L
processing chromosome A
..truncated...
In the above example the process is executed 16 times; each time a value is received from the queue channel chr_ch it is used to run the process.
Channel order
The channel guarantees that items are delivered in the same order as they have been sent, but since the process is executed in a parallel manner, there is no guarantee that they are processed in the same order as they are received.
Input files
When you need to handle files as input, you need the path qualifier. Using the path qualifier means that Nextflow will stage it in the process execution directory, and it can be accessed in the script by using the name specified in the input declaration.
The input file name can be defined dynamically by defining the input name as a Nextflow variable and referenced in the script using the $variable_name syntax.
For example, in the script below, we assign the variable name read to the input files using the path qualifier. The file is referenced using the variable substitution syntax ${read} in the script block:
//process_input_file.nf
nextflow.enable.dsl=2
process NUMLINES {
input:
path read
script:
"""
printf '${read} '
gunzip -c ${read} | wc -l
"""
}
reads_ch = Channel.fromPath( 'data/yeast/reads/ref*.fq.gz' )
workflow {
NUMLINES(reads_ch)
}
$ nextflow run process_input_file.nf -process.echo
[cd/77af6d] process > NUMLINES (1) [100%] 6 of 6 ✔
ref1_1.fq.gz 58708
ref3_2.fq.gz 52592
ref2_2.fq.gz 81720
ref2_1.fq.gz 81720
ref3_1.fq.gz 52592
ref1_2.fq.gz 58708
The input name can also be defined as a user-specified filename inside quotes.
For example, in the script below, the name of the file is specified as 'sample.fq.gz' in the input definition and can be referenced by that name in the script block.
//process_input_file_02.nf
nextflow.enable.dsl=2
process NUMLINES {
input:
path 'sample.fq.gz'
script:
"""
printf 'sample.fq.gz '
gunzip -c sample.fq.gz | wc -l
"""
}
reads_ch = Channel.fromPath( 'data/yeast/reads/ref*.fq.gz' )
workflow {
NUMLINES(reads_ch)
}
$ nextflow run process_input_file_02.nf -process.echo
[d2/eb0e9d] process > NUMLINES (1) [100%] 6 of 6 ✔
sample.fq.gz 58708
sample.fq.gz 52592
sample.fq.gz 81720
sample.fq.gz 81720
sample.fq.gz 52592
sample.fq.gz 58708
File Objects as inputs
When a process declares an input file, the corresponding channel elements must be file objects, i.e. created with the path helper function from the file specific channel factories, e.g.
Channel.fromPathorChannel.fromFilePairs.
Add input channel
For the script
process_exercise_input.nf:
- Define a Channel using
fromPathfor the transcriptomeparams.transcriptome.- Add an input channel that takes the transcriptome channel as a file input.
- Replace
params.transcriptomein thescript:block with the input variable you defined in theinput:definition.//process_exercise_input.nf nextflow.enable.dsl=2 params.chr = "A" params.transcriptome = "${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz" process CHR_COUNT { script: """ printf 'Number of sequences for chromosome '${params.chr}':' zgrep -c '^>Y'${params.chr} ${params.transcriptome} """ } workflow { CHR_COUNT() }Then run your script using
nextflow run process_exercise_input.nf -process.echoSolution
nextflow.enable.dsl=2 params.chr = "A" params.transcriptome = "${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"> > process CHR_COUNT { input: path transcriptome script: """ printf 'Number of sequences for chromosome '${params.chr}':' zgrep -c '^>Y'${params.chr} ${transcriptome} """ } transcriptome_ch = channel.fromPath(params.transcriptome) workflow { CHR_COUNT(transcriptome_ch) }N E X T F L O W ~ version 21.10.6 Launching `process_exercise_input.nf` [focused_jang] - revision: e32caf0dcb executor > local (1) [00/14ce67] process > CHR_COUNT (1) [100%] 1 of 1 ✔ Number of sequences for chromosome A:118
Combining input channels
A key feature of processes is the ability to handle inputs from multiple channels. However, it’s important to understand how the number of items within the multiple channels affect the execution of a process.
Consider the following example:
//process_combine.nf
nextflow.enable.dsl=2
process COMBINE {
input:
val x
val y
script:
"""
echo $x and $y
"""
}
num_ch = Channel.of(1, 2, 3)
letters_ch = Channel.of('a', 'b', 'c')
workflow {
COMBINE(num_ch, letters_ch)
}
$ nextflow run process_combine.nf -process.echo
Both channels contain three elements, therefore the process is executed three times, each time with a different pair:
2 and b
1 and a
3 and c
What is happening is that the process waits until it receives an input value from all the queue channels declared as input.
When this condition is verified, it uses up the input values coming from the respective queue channels, runs the task. This logic repeats until one or more queue channels have no more content. The process then stops.
What happens when not all channels have the same number of elements?
For example:
//process_combine_02.nf
nextflow.enable.dsl=2
process COMBINE {
input:
val x
val y
script:
"""
echo $x and $y
"""
}
ch_num = Channel.of(1, 2)
ch_letters = Channel.of('a', 'b', 'c', 'd')
workflow {
COMBINE(ch_num, ch_letters)
}
$ nextflow run process_combine_02.nf -process.echo
In the above example the process is executed only two times, because when a queue channel has no more data to be processed it stops the process execution.
2 and b
1 and a
Value channels and process termination
Note however that value channels, Channel.value, do not affect the process termination.
To better understand this behaviour compare the previous example with the following one:
//process_combine_03.nf
nextflow.enable.dsl=2
process COMBINE {
input:
val x
val y
script:
"""
echo $x and $y
"""
}
ch_num = Channel.value(1)
ch_letters = Channel.of('a', 'b', 'c')
workflow {
COMBINE(ch_num, ch_letters)
}
$ nextflow run process_combine_03.nf -process.echo
In this example the process is run three times.
1 and b
1 and a
1 and c
Combining input channels
Write a nextflow script
process_exercise_combine.nfthat combines two input channelstranscriptome_ch = channel.fromPath('data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz') chr_ch = channel.of('A')And include the command below in the script directive
script: """ zgrep -c ">Y${chr}" ${transcriptome} """Solution
// process_exercise_combine_answer.nf nextflow.enable.dsl=2 process COMBINE { input: path transcriptome val chr script: """ zgrep -c ">Y${chr}" ${transcriptome} """ } transcriptome_ch = channel.fromPath('data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz', checkIfExists: true) chr_ch = channel.of("A") workflow { COMBINE(transcriptome_ch, chr_ch) }
Input repeaters
We saw previously that by default the number of times a process runs is defined by the queue channel with the fewest items. However, the each qualifier allows you to repeat the execution of a process for each item in a list or a queue channel, every time new data is received.
For example if we can fix the previous example by using the input qualifer each for the letters queue channel:
//process_repeat.nf
nextflow.enable.dsl=2
process COMBINE {
input:
val x
each y
script:
"""
echo $x and $y
"""
}
ch_num = Channel.of(1, 2)
ch_letters = Channel.of('a', 'b', 'c', 'd')
workflow {
COMBINE(ch_num, ch_letters)
}
$ nextflow run process_repeat.nf -process.echo
The process will run eight times.
2 and d
1 and a
1 and c
2 and b
2 and c
1 and d
1 and b
2 and a
Input repeaters
Extend the script
process_exercise_repeat.nfby adding more values to thechrqueue channel e.g. A to P and running the process for each value.//process_exercise_repeat.nf nextflow.enable.dsl=2 process COMBINE { input: path transcriptome val chr script: """ printf "Number of sequences for chromosome $chr: " zgrep -c "^>Y${chr}" ${transcriptome} """ } transcriptome_ch = channel.fromPath('data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz', checkIfExists: true) chr_ch = channel.of('A') workflow { COMBINE(transcriptome_ch, chr_ch) }How many times does this process run?
Solution
//process_exercise_repeat_answer.nf nextflow.enable.dsl=2 process COMBINE { input: path transcriptome each chr script: """ printf "Number of sequences for chromosome $chr: " zgrep -c "^>Y${chr}" ${transcriptome} """ } transcriptome_ch = channel.fromPath('data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz', checkIfExists: true) chr_ch = channel.of('A'..'P') workflow { COMBINE(transcriptome_ch, chr_ch) }nextflow run process_exercise_repeat.nf -process.echoThis process runs 16 times.
Key Points
A Nextflow process is an independent step in a workflow
Processes contain up to five definition blocks including: directives, inputs, outputs, when clause and finally a script block.
The script block contains the commands you would like to run.
A process should have a script but the other four blocks are optional
Inputs are defined in the input block with a type qualifier and a name.
Processes Part 2
Overview
Teaching: 30 min
Exercises: 10 minQuestions
How do I get data, files, and values, out of processes?
How do I handle grouped input and output?
How can I control when a process is executed?
How do I control resources, such as number of CPUs and memory, available to processes?
How do I save output/results from a process?
Objectives
Define outputs to a process.
Understand how to handle grouped input and output using the tuple qualifier.
Understand how to use conditionals to control process execution.
Use process directives to control execution of a process.
Use the
publishDirdirective to save result files to a directory.
Outputs
We have seen how to input data into a process; now we will see how to output files and values from a process.
The output declaration block allows us to define the channels used by the process to send out the files and values produced.
An output block is not required, but if it is present it can contain one or more output declarations.
The output block follows the syntax shown below:
output:
<output qualifier> <output name>
<output qualifier> <output name>
...
Output values
Like the input, the type of output data is defined using type qualifiers.
The val qualifier allows us to output a value defined in the script.
Because Nextflow processes can only communicate through channels, if we want to share a value output of one process as input to another process, we would need to define that value in the output declaration block as shown in the following example:
//process_output_value.nf
nextflow.enable.dsl=2
params.transcriptome="${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
process COUNT_CHR_SEQS {
input:
val chr
output:
val chr
script:
"""
zgrep -c '^>Y'$chr $params.transcriptome
"""
}
// Both 'Channel' and 'channel' keywords work to generate channels.
// However, it is a good practice to be consistent through the whole pipeline development
chr_ch = channel.of('A'..'P')
workflow {
COUNT_CHR_SEQS(chr_ch)
// use the view operator to display contents of the channel
COUNT_CHR_SEQS.out.view()
}
N E X T F L O W ~ version 21.10.6
Launching `p1.nf` [jovial_lavoisier] - revision: a652ef75d4
executor > local (16)
executor > local (16)
[6a/d82669] process > COUNT_CHR_SEQS (16) [100%] 16 of 16 ✔
B
456
A
118
C
186
[..truncated..]
Output files
If we want to capture a file instead of a value as output we can use the
path qualifier that can capture one or more files produced by the process, over the specified channel.
//process_output_file.nf
nextflow.enable.dsl=2
params.transcriptome="${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
process COUNT_CHR_SEQS {
input:
val chr
output:
path "${chr}_seq_count.txt"
script:
"""
zgrep -c '^>Y'$chr $params.transcriptome > ${chr}_seq_count.txt
"""
}
// Both 'Channel' and 'channel' keywords work to generate channels.
// However, it is a good practice to be consistent through the whole pipeline development
chr_ch = channel.of('A'..'P')
workflow {
COUNT_CHR_SEQS(chr_ch)
// use the view operator to display contents of the channel
COUNT_CHR_SEQS.out.view()
}
N E X T F L O W ~ version 21.10.6
Launching `process_output_file.nf` [angry_lichterman] - revision: 6a46c69413
executor > local (16)
[95/ec5d62] process > COUNT_CHR_SEQS (13) [100%] 16 of 16 ✔
/Users/ggrimes2/Documents/process_wf/work/f2/6d5c44985a15feb0555b7b71c37a9c/J_seq_count.txt
executor > local (16)
[95/ec5d62] process > COUNT_CHR_SEQS (13) [100%] 16 of 16 ✔
work/f2/6d5c44985a15feb0555b7b71c37a9c/J_seq_count.txt
work/4f/f810942341d003acc80c2603671177/B_seq_count.txt
work/23/883ccf187b5357137a9a87d98717c0/I_seq_count.txt
[..truncated..]
In the above example the process COUNT_CHR_SEQS creates a file named <chr>_seq_count.txt in the work directory containing the number of transcripts within that chromosome.
Since a file parameter using the same name, <chr>_seq_count.txt, is declared in the output block, when the task is completed that file is sent over the output channel.
A downstream operator, such as .view or a process declaring the same channel as input will be able to receive it.
Multiple output files
When an output file name contains a * or ? metacharacter it is interpreted as a pattern match.
This allows us to capture multiple files into a list and output them as a one item channel.
For example, here we will capture the files sequence_ids.txt and sequence.txt produced as results from SPLIT_FASTA in the output channel.
//process_output_multiple.nf
nextflow.enable.dsl=2
params.transcriptome="${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
process SPLIT_FASTA {
input:
path transcriptome
output:
path "*"
script:
"""
zgrep '^>' $transcriptome > sequence_ids.txt
zgrep -v '^>' $transcriptome > sequence.txt
"""
}
// Both 'Channel' and 'channel' keywords work to generate channels.
// However, it is a good practice to be consistent through the whole pipeline development
transcriptome_ch = channel.fromPath(params.transcriptome)
workflow {
SPLIT_FASTA(transcriptome_ch)
// use the view operator to display contents of the channel
SPLIT_FASTA.out.view()
}
$ nextflow run process_output_multiple.nf
N E X T F L O W ~ version 21.10.6
Launching `process_output_multiple.nf` [goofy_meitner] - revision: 53cbf7e5a4
executor > local (1)
[21/01e6ba] process > SPLIT_FASTA (1) [100%] 1 of 1 ✔
[/work/21/01e6baac41d2f37531f86dc7a57034/sequence.txt, work/21/01e6baac41d2f37531f86dc7a57034/sequence_ids.txt]
Note: There are some caveats on glob pattern behaviour:
- Input files are not included in the list of possible matches.
- Glob pattern matches against both files and directories path.
- When a two stars pattern
**is used to recurse through subdirectories, only file paths are matched i.e. directories are not included in the result list.
Output channels
Modify the nextflow script
process_exercise_output.nfto include an output block that captures the different output file${chr}_seqids.txt.//process_exercise_output.nf nextflow.enable.dsl=2 process EXTRACT_IDS { input: path transcriptome each chr //add output block here to capture the file "${chr}_seqids.txt" script: """ zgrep '^>Y'$chr $transcriptome > ${chr}_seqids.txt """ } transcriptome_ch = channel.fromPath('data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz') chr_ch = channel.of('A'..'P') workflow { EXTRACT_IDS(transcriptome_ch, chr_ch) EXTRACT_IDS.out.view() }Solution
//process_exercise_output_answer.nf nextflow.enable.dsl=2 process EXTRACT_IDS { input: path transcriptome each chr //add output block here to capture the file "${chr}_seqids.txt" output: path "${chr}_seqids.txt" script: """ zgrep '^>Y'$chr $transcriptome > ${chr}_seqids.txt """ } transcriptome_ch = channel.fromPath('data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz') chr_ch = channel.of('A'..'P') workflow { EXTRACT_IDS(transcriptome_ch, chr_ch) EXTRACT_IDS.out.view() }
Grouped inputs and outputs
So far we have seen how to declare multiple input and output channels, but each channel was handling only one value at time. However Nextflow can handle groups of values using the tuple qualifiers.
In tuples the first item is the grouping key and the second item is the list.
[group_key,[file1,file2,...]]
When using channel containing a tuple, such a one created with .filesFromPairs factory method, the corresponding input declaration must be declared with a tuple qualifier, followed by definition of each item in the tuple.
//process_tuple_input.nf
nextflow.enable.dsl=2
process TUPLEINPUT{
input:
tuple val(sample_id), path(reads)
script:
"""
echo $sample_id
echo $reads
"""
}
reads_ch = Channel.fromFilePairs('data/yeast/reads/ref1_{1,2}.fq.gz')
workflow {
TUPLEINPUT(reads_ch)
}
outputs
ref1
ref1_1.fq.gz ref1_2.fq.gz
In the same manner an output channel containing tuple of values can be declared using the tuple qualifier following by the definition of each tuple element in the tuple.
In the code snippet below the output channel would contain a tuple with the grouping key value as the Nextflow variable sample_id and a list containing the files matching the following pattern "${sample_id}.fq.gz".
output:
tuple val(sample_id), path("${sample_id}.fq.gz")
An example can be seen in this script below.
//process_tuple_io.nf
nextflow.enable.dsl=2
process COMBINE_FQ {
input:
tuple val(sample_id), path(reads)
output:
tuple val(sample_id), path("${sample_id}.fq.gz")
script:
"""
cat $reads > ${sample_id}.fq.gz
"""
}
reads_ch = Channel.fromFilePairs('data/yeast/reads/ref1_{1,2}.fq.gz')
workflow {
COMBINE_FQ(reads_ch)
COMBINE_FQ.out.view()
}
nextflow run process_tuple_io.nf
The output is now a tuple containing the sample id and the combined fastq files.
[ref1, work/2d/a073d34b5b3231b1f57872599bd308/ref1.fq]
Composite inputs and outputs
Fill in the blank ___ input and output qualifiers for
process_exercise_tuple.nf. Note: the output for the COMBINE_REPS process.//process_exercise_tuple.nf nextflow.enable.dsl=2 process COMBINE_REPS { input: tuple ___(sample_id), ___(reads) output: tuple ___(sample_id), ___("*.fq.gz") script: """ cat *_1.fq.gz > ${sample_id}_R1.fq.gz cat *_2.fq.gz > ${sample_id}_R2.fq.gz """ } reads_ch = Channel.fromFilePairs('data/yeast/reads/ref{1,2,3}*.fq.gz',size:-1) workflow{ COMBINE_REPS(reads_ch) COMBINE_REPS.out.view() }Solution
//process_exercise_tuple_answer.nf nextflow.enable.dsl=2 process COMBINE_REPS { input: tuple val(sample_id), path(reads) output: tuple val(sample_id), path("*.fq.gz") script: """ cat *_1.fq.gz > ${sample_id}_R1.fq.gz cat *_2.fq.gz > ${sample_id}_R2.fq.gz """ } reads_ch = Channel.fromFilePairs('data/yeast/reads/ref*_{1,2,3}.fq.gz',size:-1) workflow{ COMBINE_REPS(reads_ch) COMBINE_REPS.out.view() }N E X T F L O W ~ version 21.04.0 Launching `process_exercise_tuple.nf` [spontaneous_coulomb] - revision: 06ff22f1a9 executor > local (3) [75/f4a44d] process > COMBINE_REPS (3) [100%] 3 of 3 ✔ [ref3, work/99/a7d9176e332fdc0988973dbb89df63/ref3_R1.fq.gz, work/99/a7d9176e332fdc0988973dbb89df63/ref3_R2.fq.gz] [ref2, /work/53/e3cbd39afa9f0f84a3d9cd060d991a/ref2_R1.fq.gz, /work/53/e3cbd39afa9f0f84a3d9cd060d991a/ref2_R2.fq.gz] [ref1, work/75/f4a44d0bc761fa4774c2f23a465766/ref1_R1.fq.gz, work/75/f4a44d0bc761fa4774c2f23a465766/ref1_R2.fq.gz]
Conditional execution of a process
The when declaration allows you to define a condition that must be verified in order to execute the process. This can be any expression that evaluates a boolean value; true or false.
It is useful to enable/disable the process execution depending on the state of various inputs and parameters.
In the example below the process CONDITIONAL will only execute when the value of the chr variable is less than or equal to 5:
//process_when.nf
nextflow.enable.dsl=2
process CONDITIONAL {
input:
val chr
when:
chr <= 5
script:
"""
echo $chr
"""
}
chr_ch = channel.of(1..22)
workflow {
CONDITIONAL(chr_ch)
}
4
5
2
3
1
Directives
Directive declarations allow the definition of optional settings, like the number of cpus and amount of memory, that affect the execution of the current process without affecting the task itself.
They must be entered at the top of the process body, before any other declaration blocks (i.e. input, output, etc).
Note: You do not use = when assigning a value to a directive.
Directives are commonly used to define the amount of computing resources to be used or extra information for configuration or logging purpose.
For example:
//process_directive.nf
nextflow.enable.dsl=2
process PRINTCHR {
tag "tagging with chr$chr"
cpus 1
echo true
input:
val chr
script:
"""
echo processing chromosome: $chr
echo number of cpus $task.cpus
"""
}
chr_ch = channel.of(1..22, 'X', 'Y')
workflow {
PRINTCHR(chr_ch)
}
processing chromosome: 1
number of cpus 1
processing chromosome: 2
number of cpus 1
processing chromosome: 6
number of cpus 1
[..truncated..]
The above process uses the three directives, tag, cpus and echo.
The tag directive to allow you to give a custom tag to each process execution. This tag makes it easier to identify a particular task (executed instance of a process) in a log file or in the execution report.
The second directive cpus allows you to define the number of CPUs required for each task.
The third directive echo true prints the stdout to the terminal.
We use the Nextflow task.cpus variable to capture the number of cpus assigned to a task. This is frequently used to specify the number of threads in a multi-threaded command in the script block.
Another commonly used directive is memory specification: memory.
A complete list of directives is available at this link.
Adding directives
Modify the Nextflow script
process_exercise_directives.nf
- Add a
tagdirective logging the sample_id in the execution output.- Add a
cpusdirective to specify the number of cpus as 2.- Change the fastqc
-toption value to$task.cpusin the script directive.//process_exercise_directives.nf nextflow.enable.dsl=2 process FASTQC { //add tag directive //add cpu directive input: tuple val(sample_id), path(reads) output: tuple val(sample_id), path("fastqc_out") script: """ mkdir fastqc_out fastqc $reads -o fastqc_out -t 1 """ } read_pairs_ch = Channel.fromFilePairs('data/yeast/reads/ref*_{1,2}.fq.gz') workflow { FASTQC(read_pairs_ch) FASTQC.out.view() }solution
//process_directives_answer.nf nextflow.enable.dsl=2 process FASTQC { tag "$sample_id" cpus 2 input: tuple val(sample_id), path(reads) output: tuple val(sample_id), path("fastqc_out") script: """ mkdir fastqc_out fastqc $reads -o fastqc_out -t $task.cpus """ } read_pairs_ch = Channel.fromFilePairs('data/yeast/reads/ref*_{1,2}.fq.gz') workflow { FASTQC(read_pairs_ch) FASTQC.out.view() }N E X T F L O W ~ version 21.04.0 Launching `process_exercise_directives.nf` [sad_rosalind] - revision: 2ccbfa4937 executor > local (3) [90/de1125] process > FASTQC (ref1) [100%] 3 of 3 ✔ [ref2, work/ea/9e6a341b88caf8879e8d18b77049c8/fastqc_out] [ref3, work/94/d059b816a9ec3d868f2924c26813e7/fastqc_out] [ref1, work/90/de11251d362f494d6650789d9f8c1d/fastqc_out]
Organising outputs
PublishDir directive
Nextflow manages intermediate results from the pipeline’s expected outputs independently.
Files created by a process are stored in a task specific working directory which is considered as temporary. Normally this is under the work directory, which can be deleted upon completion.
The files you want the workflow to return as results need to be defined in the process output block and then the output directory specified using the directive publishDir. More information here.
Note: A common mistake is to specify an output directory in the publishDir directive while forgetting to specify the files you want to include in the output block.
publishDir <directory>, parameter: value, parameter2: value ...
For example if we want to capture the results of the COMBINE_READS process in a results/merged_reads output directory we
need to define the files in the output and specify the location of the results directory in the publishDir directive:
//process_publishDir.nf
nextflow.enable.dsl=2
process COMBINE_READS {
publishDir "results/merged_reads"
input:
tuple val(sample_id), path(reads)
output:
path("${sample_id}.merged.fq.gz")
script:
"""
cat ${reads} > ${sample_id}.merged.fq.gz
"""
}
reads_ch = Channel.fromFilePairs('data/yeast/reads/ref1_{1,2}.fq.gz')
workflow {
COMBINE_READS(reads_ch)
}
$ nextflow run process_publishDir.nf
N E X T F L O W ~ version 21.04.0
Launching `process_publishDir.nf` [friendly_pauling] - revision: 9b5c315893
executor > local (1)
[d9/909ed9] process > COMBINE_READS (1) [100%] 1 of 1 ✔
We can use the UNIX command tree to examine the contents of the results directory.
tree results
results/
└── merged_reads
└── ref1.merged.fq.gz -> work/48/f97234d7185cbfbd86e2f11c1afab5/ref1.merged.fq.gz
In the above example, the publishDir "results/merged_reads", creates a symbolic link -> to the output files specified by the process merged_reads to the directory path results/merged_reads.
publishDir
The publishDir output is relative to the path the pipeline run has been launched. Hence, it is a good practice to use implicit variables like
projectDirto specify publishDir value.
publishDir parameters
The publishDir directive can take optional parameters, for example the mode parameter can take the value "copy" to specify that you wish to copy the file to output directory rather than just a symbolic link to the files in the working directory. Since the working directory is generally deleted on completion of a pipeline, it is safest to use mode: "copy" for results files. The default mode (symlink) is helpful for checking intermediate files which are not needed in the long term.
publishDir "results/merged_reads", mode: "copy"
Full list here.
Manage semantic sub-directories
You can use more than one publishDir to keep different outputs in separate directories. To specify which files to put in which output directory use the parameter pattern with the a glob pattern that selects which files to publish from the overall set of output files.
In the example below we will create an output folder structure in the directory results, which contains a separate sub-directory for sequence id file, pattern:"*_ids.txt" , and a sequence directory, results/sequence" for the sequence.txt file. Remember, we need to specify the files we want to copy as outputs.
//process_publishDir_semantic.nf
nextflow.enable.dsl=2
params.transcriptome="${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
process SPLIT_FASTA {
publishDir "results/ids", pattern: "*_ids.txt", mode: "copy"
publishDir "results/sequence", pattern: "sequence.txt", mode: "copy"
input:
path transcriptome
output:
path "*"
script:
"""
zgrep '^>' $transcriptome > sequence_ids.txt
zgrep -v '^>' $transcriptome > sequence.txt
"""
}
// Both 'Channel' and 'channel' keywords work to generate channels.
// However, it is a good practice to be consistent through the whole pipeline development
transcriptome_ch = channel.fromPath(params.transcriptome)
workflow {
SPLIT_FASTA(transcriptome_ch)
// use the view operator to display contents of the channel
SPLIT_FASTA.out.view()
}
$ nextflow run process_publishDir_semantic.nf
N E X T F L O W ~ version 21.04.0
Launching `process_publishDir_semantic.nf` [golden_poisson] - revision: 421a604840
executor > local (1)
[be/950786] process > SPLIT_FASTA (1) [100%] 1 of 1 ✔
We can now use the tree command to examine the results directory.
$ tree results
results/
├── sequence
│ └── sequence.txt
└── ids
└── sequence_ids.txt
Publishing results
Add a
publishDirdirective to the nextflow scriptprocess_exercise_publishDir.nfthat copies the merged reads to the results folder merged_reps.//process_exercise_publishDir.nf nextflow.enable.dsl=2 params.reads= "data/yeast/reads/ref{1,2,3}*{1,2}.fq.gz" process MERGE_REPS { input: tuple val(sample_id), path(reads) output: path("*fq.gz") script: """ cat *1.fq.gz > ${sample_id}.merged.R1.fq.gz cat *2.fq.gz > ${sample_id}.merged.R2.fq.gz """ } reads_ch = Channel.fromFilePairs(params.reads,checkIfExists:true,size:6) workflow { MERGE_REPS(reads_ch) }Solution
//process_exercise_publishDir_answer.nf nextflow.enable.dsl=2 params.reads= "data/yeast/reads/ref{1,2,3}*{1,2}.fq.gz" process MERGE_REPS { publishDir "results/merged_reps" input: tuple val(sample_id), path(reads) output: path("*fq.gz") script: """ cat *1.fq.gz > ${sample_id}.merged.R1.fq.gz cat *2.fq.gz > ${sample_id}.merged.R2.fq.gz """ } reads_ch = Channel.fromFilePairs(params.reads,checkIfExists:true,size:6) workflow { MERGE_REPS(reads_ch) }$ nextflow run process_exercise_publishDir.nfN E X T F L O W ~ version 21.04.0 Launching `process_exercise_publishDir.nf` [infallible_becquerel] - revision: 4d865241a8 executor > local (1) [22/88aa22] process > MERGE_REPS (1) [100%] 1 of 1 ✔
Nextflow Patterns
If you want to find out common structures of Nextflow processes, the Nextflow Patterns page collects some recurrent implementation patterns used in Nextflow applications.
Key Points
Outputs to a process are defined using the output blocks.
You can group input and output data from a process using the tuple qualifier.
The execution of a process can be controlled using the
whendeclaration and conditional statements.Files produced within a process and defined as
outputcan be saved to a directory using thepublishDirdirective.
Workflow
Overview
Teaching: 20 min
Exercises: 20 minQuestions
How do I connect channels and processes to create a workflow?
How do I invoke a process inside a workflow?
Objectives
Create a Nextflow workflow joining multiple processes.
Understand how to to connect processes via their inputs and outputs within a workflow.
Workflow
Our previous episodes have shown us how to parameterise workflows using params, move data around a workflow using channels and define individual tasks using processes. In this episode we will cover how connect multiple processes to create a workflow.
Workflow definition
We can connect processes to create our pipeline inside a workflow scope.
The workflow scope starts with the keyword workflow, followed by an optional name and finally the workflow body delimited by curly brackets {}.
Implicit workflow
A workflow definition which does not declare any name is assumed to be the main workflow, and it is implicitly executed. Therefore it’s the entry point of the workflow application.
Invoking processes with a workflow
As seen previously, a process is invoked as a function in the workflow scope, passing the expected input channels as arguments as it if were.
<process_name>(<input_ch1>,<input_ch2>,...)
To combined multiple processes invoke them in the order they would appear in a workflow. When invoking a process with multiple inputs, provide them in the same order in which they are declared in the input block of the process.
For example:
//workflow_01.nf
nextflow.enable.dsl=2
process FASTQC {
input:
tuple(val(sample_id), path(reads))
output:
path "fastqc_${sample_id}_logs"
script:
"""
mkdir fastqc_${sample_id}_logs
fastqc -o fastqc_${sample_id}_logs -f fastq -q ${reads}
"""
}
process MULTIQC {
publishDir "results/mqc"
input:
path transcriptome
output:
path "*"
script:
"""
multiqc .
"""
}
workflow {
read_pairs_ch = channel.fromFilePairs('data/yeast/reads/*_{1,2}.fq.gz',checkIfExists: true)
//index process takes 1 input channel as a argument
//assign process output to Nextflow variable fastqc_obj
fastqc_obj = FASTQC(read_pairs_ch)
//quant channel takes 1 input channel as an argument
//We use the collect operator to gather multiple channel items into a single item
MULTIQC(fastqc_obj.collect()).view()
}
Process outputs
In the previous example we assigned the process output to a Nextflow variable fastqc_obj.
A process output can also be accessed directly using the out attribute for the respective process object.
For example:
[..truncated..]
workflow {
read_pairs_ch = channel.fromFilePairs('data/yeast/reads/*_{1,2}.fq.gz',checkIfExists: true)
FASTQC(read_pairs_ch)
// process output accessed using the `out` attribute of the process object
MULTIQC(FASTQC.out.collect()).view()
MULTIQC.out.view()
}
When a process defines two or more output channels, each of them can be accessed using the list element operator e.g. out[0], out[1], or using named outputs.
Process named output
It can be useful to name the output of a process, especially if there are multiple outputs.
The process output definition allows the use of the emit: option to define a named identifier that can be used to reference the channel in the external scope.
For example in the script below we name the output from the FASTQC process as fastqc_results using the emit: option. We can then reference the output as
FASTQC.out.fastqc_results in the workflow scope.
//workflow_02.nf
nextflow.enable.dsl=2
process FASTQC {
input:
tuple val(sample_id), path(reads)
output:
path "fastqc_${sample_id}_logs", emit: fastqc_results
script:
"""
mkdir fastqc_${sample_id}_logs
fastqc -o fastqc_${sample_id}_logs ${reads}
"""
}
process MULTIQC {
publishDir "results/mqc"
input:
path fastqc_results
output:
path "*"
script:
"""
multiqc .
"""
}
workflow {
read_pairs_ch = channel.fromFilePairs('data/yeast/reads/ref*_{1,2}.fq.gz',checkIfExists: true)
//FASTQC process takes 1 input channel as a argument
FASTQC(read_pairs_ch)
//MULTIQC channel takes 1 input channels as arguments
MULTIQC(FASTQC.out.fastqc_results.collect()).view()
}
Accessing script parameters
A workflow component can access any variable and parameter defined in the outer scope:
For example:
//workflow_03.nf
[..truncated..]
params.reads = 'data/yeast/reads/*_{1,2}.fq.gz'
workflow {
reads_ch_ = channel.fromFilePairs(params.reads)
FASTQC(reads_ch_)
MULTIQC(FASTQC.out.fastqc_results.collect()).view()
}
In this example params.reads, defined outside the workflow scope, can be accessed inside the workflow scope.
Workflow
Connect the output of the process
FASTQCtoPARSEZIPin the Nextflow scriptworkflow_exercise.nf.Note: You will need to pass the
read_pairs_chas an argument to FASTQC and you will need to use thecollectoperator to gather the items in the FASTQC channel output to a single List item.//workflow_exercise.nf nextflow.enable.dsl=2 params.reads = 'data/yeast/reads/*_{1,2}.fq.gz' process FASTQC { input: tuple val(sample_id), path(reads) output: path "fastqc_${sample_id}_logs/*.zip" script: """ mkdir fastqc_${sample_id}_logs fastqc -o fastqc_${sample_id}_logs ${reads} """ } process PARSEZIP { publishDir "results/fqpass", mode:"copy" input: path fastqc_logs output: path 'pass_basic.txt' script: """ for zip in *.zip; do zipgrep 'Basic Statistics' \$zip|grep 'summary.txt'; done > pass_basic.txt """ } read_pairs_ch = channel.fromFilePairs(params.reads,checkIfExists: true) workflow { //connect process FASTQC and PARSEZIP // remember to use the collect operator on the FASTQC output }Solution
//workflow_exercise.nf nextflow.enable.dsl=2 params.reads = 'data/yeast/reads/*_{1,2}.fq.gz' process FASTQC { input: tuple val(sample_id), path(reads) output: path "fastqc_${sample_id}_logs/*.zip" script: """ mkdir fastqc_${sample_id}_logs fastqc -o fastqc_${sample_id}_logs ${reads} """ } process PARSEZIP { publishDir "results/fqpass", mode:"copy" input: path fastqc_logs output: path 'pass_basic.txt' script: """ for zip in *.zip; do zipgrep 'Basic Statistics' \$zip|grep 'summary.txt'; done > pass_basic.txt """ } read_pairs_ch = channel.fromFilePairs(params.reads,checkIfExists: true) workflow { PARSEZIP(FASTQC(read_pairs_ch).collect()) }$ nextflow run workflow_exercise.nf$ wc -l results/fqpass/pass_basic.txt18The file
results/fqpass/pass_basic.txtshould have 18 lines. If you only have two lines it might mean that you did not usecollect()operator on the FASTC output channel.
Key Points
A Nextflow workflow is defined by invoking
processesinside theworkflowscope.A process is invoked like a function inside the
workflowscope passing any required input parameters as arguments. e.g.FASTQC(reads_ch).Process outputs can be accessed using the
outattribute for the respectiveprocessobject or assigning the output to a Nextflow variable. - Multiple outputs from a single process can be accessed using the list syntax[]and it’s index or by referencing the a named process output .
Operators
Overview
Teaching: 30 min
Exercises: 10 minQuestions
How do I perform operations, such as filtering, on channels?
What are the different kinds of operations I can perform on channels?
How do I combine operations?
How can I use a CSV file to process data into a Channel?
Objectives
Understand what Nextflow operators are.
Modify the contents/elements of a channel using operators.
Perform filtering and combining operations on a channel object.
Use the
splitCsvoperator to parse the contents of CSV file into a channel .
Operators
In the Channels episode we learnt how to create Nextflow channels to enable us to pass data and values around our workflow. If we want to modify the contents or behaviour of a channel, Nextflow provides methods called operators. We have previously used the view operator to view the contents of a channel. There are many more operator methods that can be applied to Nextflow channels that can be usefully separated into several groups:
- Filtering operators: reduce the number of elements in a channel.
- Transforming operators: transform the value/data in a channel.
- Splitting operators: split items in a channel into smaller chunks.
- Combining operators: join channel together.
- Forking operators: split a single channel into multiple channels.
- Maths operators: apply simple math function on channels.
- Other: such as the view operator.
In this episode you will see examples, and get to use different types of operators.
Using Operators
To use an operator, the syntax is the channel name, followed by a dot . , followed by the operator name and brackets ().
channel_obj.<operator>()
view
The view operator prints the items emitted by a channel to the console appending a new line character to each item in the channel.
ch = channel.of('1', '2', '3')
ch.view()
We can also chain together the channel factory method .of and the operator .view() using the
dot notation.
ch = channel.of('1', '2', '3').view()
To make code more readable we can split the operators over several lines. The blank space between the operators is ignored and is solely for readability.
ch = channel
.of('1', '2', '3')
.view()
prints:
1
2
3
Closures
An optional closure {} parameter can be specified to customise how items are printed.
Briefly, a closure is a block of code that can be passed as an argument to a function. In this way you can define a chunk of code and then pass it around as if it were a string or an integer. By default the parameters for a closure are specified with the groovy keyword $it (‘it’ is for ‘item’).
For example here we use the the view operator and apply a closure to it, to add a chr prefix to each element of the channel using string interpolation.
ch = channel
.of('1', '2', '3')
.view({ "chr$it" })
It prints:
chr1
chr2
chr3
Note: the view() operator doesn’t change the contents of the channel object.
ch = channel
.of('1', '2', '3')
.view({ "chr$it" })
ch.view()
chr1
chr2
chr3
1
2
3
Filtering operators
We can reduce the number of items in a channel by using filtering operators.
The filter operator allows you to get only the items emitted by a channel that satisfy a condition and discard all the others. The filtering condition can be specified by using either:
- a regular expression
- a literal value
- a data type qualifier, e.g. Number (any integer,float …), String, Boolean
- or any boolean statement.
Data type qualifier
Here we use the filter operator on the chr_ch channel specifying the data type qualifier Number so that only numeric items are returned. The Number data type includes both integers and floating point numbers.
We will then use the view operator to print the contents.
chr_ch = channel.of( 1..22, 'X', 'Y' )
autosomes_ch =chr_ch.filter( Number )
autosomes_ch.view()
To simplify the code we can chain multiple operators together, such as filter and view using a . .
The previous example could be rewritten like: The blank space between the operators is ignored and is used for readability.
chr_ch = channel
.of( 1..22, 'X', 'Y' )
.filter( Number )
.view()
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
Regular expression
To filter by a regular expression you have to do is to put ~ right in front of the string literal regular expression (e.g. ~"(^[Nn]extflow)" or use slashy strings which replace the quotes with /. ~/^[Nn]extflow/).
The following example shows how to filter a channel by using a regular expression ~/^1.*/ inside a slashy string, that returns only strings that begin with 1:
chr_ch = channel
.of( 1..22, 'X', 'Y' )
.filter(~/^1.*/)
.view()
1
10
11
12
13
14
15
16
17
18
19
Boolean statement
A filtering condition can be defined by using a Boolean expression described by a closure {} and returning a boolean value. For example the following fragment shows how to combine a filter for a type qualifier Number with another filter operator using a Boolean expression to emit numbers less than 5:
channel
.of( 1..22, 'X', 'Y' )
.filter(Number)
.filter { it < 5 }
.view()
1
2
3
4
Closures
In the above example we could remove the brackets around the filter condition e.g.
filter{ it<5}, since it specifies a closure as the operator’s argument. This is language short forfilter({ it<5})
Literal value
Finally, if we only want to include elements of a specific value we can specify a literal value. In the example below we use the literal value X to filter the channel for only those elements containing the value X.
channel
.of( 1..22, 'X', 'Y' )
.filter('X')
.view()
X
Filter a channel
Add two channel filters to the Nextflow script below to view only the even numbered chromosomes.
Note: The expression
it % 2produces the remainder of a division.chr_ch = channel .of( 1..22, 'X', 'Y' ) .view()Solution
chr_ch = channel .of( 1..22, 'X', 'Y' ) .filter( Number ) .filter({ it % 2 == 0 }) .view()2 4 6 8 10 12 14 16 18 20 22
Modifying the contents of a channel
If we want to modify the items in a channel, we can use transforming operators.
map
Applying a function to items in a channel
The map operator applies a function of your choosing to every item in a channel, and returns the items so obtained as a new channel. The function applied is called the mapping function and is expressed with a closure {} as shown in the example below:
chr = channel
.of( 'chr1', 'chr2' )
.map ({ it.replaceAll("chr","") })
chr.view()
Here the map function uses the groovy string function replaceAll to remove the chr prefix from each element.
1
2
We can also use the map operator to transform each element into a tuple.
In the example below we use the map operator to transform a channel containing fastq files to a new channel containing a tuple with the fastq file and the number of reads in the fastq file. We use the built in countFastq file method to count the number of records in a FASTQ formatted file.
We can change the default name of the closure parameter keyword from it to a more meaningful name file using ->. When we have multiple parameters we can specify the keywords at the start of the closure, e.g. file, numreads ->.
fq_ch = channel
.fromPath( 'data/yeast/reads/*.fq.gz' )
.map ({ file -> [file, file.countFastq()] })
.view ({ file, numreads -> "file $file contains $numreads reads" })
This would produce.
file data/yeast/reads/ref1_2.fq.gz contains 14677 reads
file data/yeast/reads/etoh60_3_2.fq.gz contains 26254 reads
file data/yeast/reads/temp33_1_2.fq.gz contains 20593 reads
file data/yeast/reads/temp33_2_1.fq.gz contains 15779 reads
file data/yeast/reads/ref2_1.fq.gz contains 20430 reads
[..truncated..]
We can then add a filter operator to only retain those fastq files with more than 25000 reads.
channel
.fromPath( 'data/yeast/reads/*.fq.gz' )
.map ({ file -> [file, file.countFastq()] })
.filter({ file, numreads -> numreads > 25000})
.view ({ file, numreads -> "file $file contains $numreads reads" })
file data/yeast/reads/etoh60_3_2.fq.gz contains 26254 reads
file data/yeast/reads/etoh60_3_1.fq.gz contains 26254 reads
map operator
Add a
mapoperator to the Nextflow script below to transform the contents into a tuple with the file and the file’s name, using the.getNamemethod. ThegetNamemethod gives the filename. Finallyviewthe channel contents.channel .fromPath( 'data/yeast/reads/*.fq.gz' ) .view()Solution
ch = channel .fromPath( 'data/yeast/reads/*.fq.gz' ) .map ({file -> [ file, file.getName() ]}) .view({file, name -> "file's name: $name"})
Converting a list into multiple items
The flatten operator transforms a channel in such a way that every item in a list or tuple is flattened so that each single entry is emitted as a sole element by the resulting channel.
list1 = [1,2,3]
ch = channel
.of(list1)
.view()
[1, 2, 3]
ch =channel
.of(list1)
.flatten()
.view()
The above snippet prints:
1
2
3
This is similar to the channel factory Channel.fromList.
Converting the contents of a channel to a single list item.
The reverse of the flatten operator is collect. The collect operator collects all the items emitted by a channel to a list and return the resulting object as a sole emission. This can be extremely useful when combining the results from the output of multiple processes, or a single process run multiple times.
ch = channel
.of( 1, 2, 3, 4 )
.collect()
.view()
It prints a single value:
[1,2,3,4]
The result of the collect operator is a value channel and can be used multiple times.
Grouping contents of a channel by a key.
The groupTuple operator collects tuples or lists of values by grouping together the channel elements that share the same key. Finally it emits a new tuple object for each distinct key collected.
For example.
ch = channel
.of( ['wt','wt_1.fq'], ['wt','wt_2.fq'], ["mut",'mut_1.fq'], ['mut', 'mut_2.fq'] )
.groupTuple()
.view()
[wt, [wt_1.fq, wt_1.fq]]
[mut, [mut_1.fq, mut_2.fq]]
If we know the number of items to be grouped we can use the groupTuple size parameter.
When the specified size is reached, the tuple is emitted. By default incomplete tuples (i.e. with less than size grouped items) are discarded (default).
For example.
ch = channel
.of( ['wt','wt_1.fq'], ['wt','wt_1.fq'], ["mut",'mut_1.fq'])
.groupTuple(size:2)
.view()
outputs,
[wt, [wt_1.fq, wt_1.fq]]
This operator is useful to process altogether all elements for which there’s a common property or a grouping key.
Group Tuple
channel.fromPath('data/yeast/reads/*.fq.gz') .view()Modify the Nextflow script above to add the
mapoperator to create a tuple with the name prefix as the key and the file as the value using the closure below.{ file -> [ file.getName().split('_')[0], file ] }Finally group together all files having the same common prefix using the
groupTupleoperator andviewthe contents of the channel.Solution
ch = channel.fromPath('data/yeast/reads/*.fq.gz') .map { file -> [ file.getName().split('_')[0], file ] } .groupTuple() .view()
Merging Channels
Combining operators allows you to merge channels together. This can be useful when you want to combine the output channels from multiple processes to perform another task such as joint QC.
mix
The mix operator combines the items emitted by two (or more) channels into a single channel.
ch1 = channel.of( 1,2,3 )
ch2 = channel.of( 'X','Y' )
ch3 = channel.of( 'mt' )
ch4 = ch1.mix(ch2,ch3).view()
1
2
3
X
Y
mt
The items emitted by the resulting mixed channel may appear in any order, regardless of which source channel they came from. Thus, the following example it could be a possible result of the above example as well.
1
2
X
3
mt
Y
join
The join operator creates a channel that joins together the items emitted by two channels for which exists a matching key. The key is defined, by default, as the first element in each item emitted.
reads1_ch = channel
.of(['wt', 'wt_1.fq'], ['mut','mut_1.fq'])
reads2_ch= channel
.of(['wt', 'wt_2.fq'], ['mut','mut_2.fq'])
reads_ch = reads1_ch
.join(reads2_ch)
.view()
The resulting channel emits:
[wt, wt_1.fq, wt_2.fq]
[mut, mut_1.fq, mut_2.fq]
Forking operators
Forking operators split a single channel into multiple channels.
into
The into operator connects a source channel to two or more target channels in such a way the values emitted by the source channel are copied to the target channels. Channel names are separated by a semi colon. For example:
channel
.of( 'chr1', 'chr2', 'chr3' )
.into({ ch1; ch2 })
ch1.view({"ch1 emits: $it"})
ch2.view({"ch2 emits: $it"})
Produces.
ch1 emits: chr1
ch1 emits: chr2
ch2 emits: chr1
ch1 emits: chr3
ch2 emits: chr2
ch2 emits: chr3
Maths operators
The maths operators allows you to apply simple math function on channels.
The maths operators are:
- count
- min
- max
- sum
- toInteger
Counting items in a channel
The count operator creates a channel that emits a single item: a number that represents the total number of items emitted by the source channel. For example:
ch = channel
.of(1..22,'X','Y')
.count()
.view()
24
Splitting items in a channel
Sometimes you want to split the content of a individual item in a channel, like a file or string, into smaller chunks that can be processed by downstream operators or processes e.g. items stored in a CSV file.
Nextflow has a number of splitting operators that can achieve this:
- splitCsv: The splitCsv operator allows you to parse text items emitted by a channel, that are formatted using the CSV format, and split them into records or group them into list of records with a specified length.
- splitFasta: The splitFasta operator allows you to split the entries emitted by a channel, that are formatted using the FASTA format. It returns a channel which emits a text item for each sequence in the received FASTA content.
- splitFastq: The splitFastq operator allows you to split the entries emitted by a channel, that are formatted using the FASTQ format. It returns a channel which emits a text chunk for each sequence in the received item.
- splitText: The splitText operator allows you to split multi-line strings or text file items, emitted by a source channel into chunks containing n lines, which will be emitted by the resulting channel.
splitCsv
The splitCsv operator allows you to parse text items emitted by a channel, that are formatted using the CSV format, and split them into records or group them into list of records with a specified length. This is useful when you want to use a sample sheet.
In the simplest case just apply the splitCsv operator to a channel emitting a CSV formatted text files or text entries. For example:
For the CSV file samples.csv.
cat data/yeast/samples.csv
sample_id,fastq_1,fastq_2
ref1,data/yeast/reads/ref1_1.fq.gz,data/yeast/reads/ref1_2.fq.gz
ref2,data/yeast/reads/ref2_1.fq.gz,data/yeast/reads/ref2_2.fq.gz
We can use the splitCsv() operator to split the channel contaning a CSV file into three elements.
csv_ch=channel
.fromPath('data/yeast/samples.csv')
.splitCsv()
csv_ch.view()
[sample_id, fastq_1, fastq_2]
[ref1, data/yeast/reads/ref1_1.fq.gz, data/yeast/reads/ref1_2.fq.gz]
[ref2, data/yeast/reads/ref2_1.fq.gz, data/yeast/reads/ref2_2.fq.gz]
The above example shows hows the CSV file samples.csv is parsed and is split into three elements.
Accessing values
Values can be accessed by their positional indexes using the square brackets syntax[index]. So to access the first column you would use [0] as shown in the following example:
csv_ch=channel
.fromPath('data/yeast/samples.csv')
.splitCsv()
csv_ch
.view({it[0]})
sample_id
ref1
ref2
Column headers
When the CSV begins with a header line defining the column names, you can specify the parameter header: true which allows you to reference each value by its name, as shown in the following example:
csv_ch=channel
.fromPath('data/yeast/samples.csv')
.splitCsv(header:true)
csv_ch.view({it.fastq_1})
data/yeast/reads/ref1_1.fq.gz
data/yeast/reads/ref2_1.fq.gz
Parse a CSV file
Modify the Nextflow script to print the first column
sample_id.csv_ch=channel .fromPath('data/yeast/samples.csv')Solution
csv_ch=channel .fromPath('data/yeast/samples.csv') .splitCsv(header:true) csv_ch.view({it.sample_id})
Tab delimited files
If you want to split a tab delimited file or file separated by another character use the sep parameter of the split splitCsv operator.
For examples,
Channel.of("val1\tval2\tval3\nval4\tval5\tval6\n")
.splitCsv(sep: "\t")
.view()
[val1, val2, val3]
[val4, val5, val6]
More resources
See the operators documentation on the Nextflow web site.
Key Points
Nextflow operators are methods that allow you to modify, set or view channels.
Operators can be separated in to several groups; filtering , transforming , splitting , combining , forking and Maths operators
To use an operator use the dot notation after the Channel object e.g.
my_ch.view().You can parse text items emitted by a channel, that are formatted using the CSV format, using the
splitCsvoperator.
Nextflow configuration
Overview
Teaching: 30 min
Exercises: 15 minQuestions
What is the difference between the workflow implementation and the workflow configuration?
How do I configure a Nextflow workflow?
How do I assign different resources to different processes?
How do I separate and provide configuration for different computational systems?
How do I change configuration settings from the default settings provided by the workflow?
Objectives
Understand the difference between workflow implementation and configuration.
Understand the difference between configuring Nextflow and a Nextflow script.
Create a Nextflow configuration file.
Understand what a configuration scope is.
Be able to assign resources to a process.
Be able to refine configuration settings using process selectors.
Be able to group configurations into profiles for use with different computer infrastructures.
Be able to override existing settings.
Be able to inspect configuration settings before running a workflow.
Nextflow configuration
A key Nextflow feature is the ability to decouple the workflow implementation, which describes the flow of data and operations to perform on that data, from the configuration settings required by the underlying execution platform. This enables the workflow to be portable, allowing it to run on different computational platforms such as an institutional HPC or cloud infrastructure, without needing to modify the workflow implementation.
We have seen earlier that it is possible to provide a process with
directives. These directives are process specific configuration settings.
Similarly, we have also provided parameters to our workflow which
are parameter configuration settings. These configuration settings
can be separated from the workflow implementation, into a
configuration file.
Configuration files
Settings in a configuration file are sets of name-value pairs
(name = value). The name is a specific property to set,
while the value can be anything you can assign to a variable (see
nextflow scripting), for example, strings,
booleans, or other variables.
It is also possible to access any variable defined in the
host environment such as $PATH, $HOME, $PWD, etc.
// nextflow.config
my_home_dir = "$HOME"
Accessing variables in your configuration file
Generally, variables and functions defined in a configuration file are not accessible from the workflow script. Only variables defined using the
paramsscope and theenvscope (withoutenvprefix) can be accessed from the workflow script.workflow { MY_PROCESS( params.input ) }
Settings are also partitioned into
scopes, which govern the behaviour of different elements of the
workflow. For example, workflow parameters are governed from the
params scope, while process directives are governed from the process scope. A full list of the available scopes can be found in the
documentation. It is also possible to define your own scope.
Configuration settings for a workflow are often stored in the file
nextflow.config which is in the same directory as the workflow script.
Configuration can be written in either of two ways. The first is using
dot notation, and the second is using brace notation. Both forms
of notation can be used in the same configuration file.
An example of dot notation:
params.input = '' // The workflow parameter "input" is assigned an empty string to use as a default value
params.outdir = './results' // The workflow parameter "outdir" is assigned the value './results' to use by default.
An example of brace notation:
params {
input = ''
outdir = './results'
}
Configuration files can also be separated into multiple files and
included into another using the includeConfig statement.
// nextflow.config
params {
input = ''
outdir = './results'
}
includeConfig 'system_resources.config'
// system_resources.config
process {
cpus = 1 // default cpu usage
time = '1h' // default time limit
}
How configuration files are combined
Configuration settings can be spread across several files. This also allows settings to be overridden by other configuration files. The priority of a setting is determined by the following order, ranked from highest to lowest.
- Parameters specified on the command line (
--param_name value). - Parameters provided using the
-params-fileoption. - Config file specified using the
-cmy_config option. - The config file named
nextflow.configin the current directory. - The config file named
nextflow.configin the workflow project directory ($projectDir: the directory where the script to be run is located). - The config file
$HOME/.nextflow/config. - Values defined within the workflow script itself (e.g.,
main.nf).
If configuration is provided by more than one of these methods, configuration is merged giving higher priority to configuration provided higher in the list.
Existing configuration can be completely ignored by using -C <custom.config> to use only configuration provided in the custom.config file.
Configuring Nextflow vs Configuring a Nextflow workflow
Parameters starting with a single dash
-(e.g.,-c my_config.config) are configuration options fornextflow, while parameters starting with a double dash--(e.g.,--outdir) are workflow parameters defined in theparamsscope.The majority of Nextflow configuration settings must be provided on the command-line, however a handful of settings can also be provided within a configuration file, such as
workdir = '/path/to/work/dir'(-w /path/to/work/dir) orresume = true(-resume), and do not belong to a configuration scope.
Configuring Nextflow vs Configuring a Nextflow workflow
Parameters starting with a single dash
-(e.g.,-c my_config.config) are configuration options fornextflow, while parameters starting with a double dash--(e.g.,--outdir) are workflow parameters defined in theparamsscope.The majority of Nextflow configuration settings must be provided on the command-line, however a handful of settings can also be provided within a configuration file, such as
workdir = '/path/to/work/dir'(-w /path/to/work/dir) orresume = true(-resume), and do not belong to a configuration scope.
Determine script output
Determine the outcome of the following script executions. Given the script
print_message.nf:nextflow.enable.dsl = 2 params.message = 'hello' workflow { PRINT_MESSAGE(params.message) } process PRINT_MESSAGE { echo true input: val my_message script: """ echo $my_message """ }and configuration (
print_message.config):params.message = 'Are you tired?'What is the outcome of the following commands?
nextflow run print_message.nfnextflow run print_message.nf --message '¿Que tal?'nextflow run print_message.nf -c print_message.confignextflow run print_message.nf -c pring_message.config --message '¿Que tal?'Solution
- ‘hello’ - Workflow script uses the value in
print_message.nf- ‘¿Que tal?’ - The command-line parameter overrides the script setting.
- ‘Are you tired?’ - The configuration overrides the script setting
- ‘¿Que tal?’ - The command-line parameter overrides both the script and configuration settings.
Configuring process behaviour
Earlier we saw that process directives allow the specification of
settings for the task execution such as cpus, memory, conda
and other resources in the pipeline script. This is useful when
prototyping a small workflow script, however this ties the configuration
to the workflow, making it less portable. A good practice is to
separate the process configuration settings into another file.
The process configuration scope allows the setting of any process directives in the Nextflow configuration file.
For example:
// nextflow.config
process {
cpus = 2
memory = 8.GB
time = '1 hour'
publishDir = [ path: params.outdir, mode: 'copy' ]
}
Unit values
Memory and time duration units can be specified either using a string based notation in which the digit(s) and the unit can be separated by a space character, or by using the numeric notation in which the digit(s) and the unit are separated by a dot character and not enclosed by quote characters.
String syntax Numeric syntax Value ‘10 KB’ 10.KB 10240 bytes ‘500 MB’ 500.MB 524288000 bytes ‘1 min’ 1.min 60 seconds ‘1 hour 25 sec’ - 1 hour and 25 seconds
These settings are applied to all processes in the workflow. A process selector can be used to apply the configuration to a specific process or group of processes.
Process selectors
The resources for a specific process can be defined using withName:
followed by the process name ( either the simple name e.g., 'FASTQC',
or the fully qualified name e.g., 'NFCORE_RNASEQ:RNA_SEQ:SAMTOOLS_SORT'),
and the directives within curly braces.
For example, we can specify different cpus and memory resources
for the processes INDEX and FASTQC as follows:
// process_resources.config
process {
withName: INDEX {
cpus = 4
memory = 8.GB
}
withName: FASTQC {
cpus = 2
memory = 4.GB
}
}
When a workflow has many processes, it is inconvenient to specify
directives for all processes individually, especially if directives
are repeated for groups of processes. A helpful strategy is to annotate
the processes using the label directive (processes can have multiple
labels). The withLabel selector then allows the configuration of all
processes annotated with a specific label, as shown below:
// configuration_process_labels.nf
nextflow.enable.dsl=2
process P1 {
label "big_mem"
script:
"""
echo P1: Using $task.cpus cpus and $task.memory memory.
"""
}
process P2 {
label "big_mem"
script:
"""
echo P2: Using $task.cpus cpus and $task.memory memory.
"""
}
workflow {
P1()
P2()
}
// configuration_process-labels.config
process {
withLabel: big_mem {
cpus = 16
memory = 64.GB
}
}
Another strategy is to use process selector expressions. Both
withName: and withLabel: allow the use of regular expressions
to apply the same configuration to all processes matching a pattern.
Regular expressions must be quoted, unlike simple process names
or labels.
- The
|matches either-or, e.g.,withName: 'INDEX|FASTQC'applies the configuration to any process matching the nameINDEXorFASTQC. - The
!inverts a selector, e.g.,withLabel: '!small_mem'applies the configuration to any process without thesmall_memlabel. - The
.*matches any number of characters, e.g.,withName: 'NFCORE_RNASEQ:RNA_SEQ:BAM_SORT:.*'matches all processes of the workflowNFCORE_RNASEQ:RNA_SEQ:BAM_SORT.
A regular expression cheat-sheet can be found here if you would like to write more expressive expressions.
Selector priority
When mixing generic process configuration and selectors, the following priority rules are applied (from highest to lowest):
withNameselector definition.withLabelselector definition.- Process specific directive defined in the workflow script.
- Process generic
processconfiguration.
Process selectors
Create a Nextflow config,
process-selector.config, specifying differentcpusandmemoryresources for the two processesP1(cpus 1 and memory 2.GB) andP2(cpus 2 and memory 1.GB), whereP1andP2are defined as follows:// process-selector.nf nextflow.enable.dsl=2 process P1 { echo true script: """ echo P1: Using $task.cpus cpus and $task.memory memory. """ } process P2 { echo true script: """ echo P2: Using $task.cpus cpus and $task.memory memory. """ } workflow { P1() P2() }Solution
// process-selector.config process { withName: P1 { cpus = 1 memory = 2.GB } withName: P2 { cpus = 2 memory = 1.GB } }$ nextflow run process-selector.nf -c process-selector.config -process.echoN E X T F L O W ~ version 21.04.0 Launching `process-selector.nf` [clever_borg] - revision: e765b9e62d executor > local (2) [de/86cef0] process > P1 [100%] 1 of 1 ✔ [bf/8b332e] process > P2 [100%] 1 of 1 ✔ P2: Using 2 cpus and 1 GB memory. P1: Using 1 cpus and 2 GB memory.
Dynamic expressions
A common scenario is that configuration settings may depend on the
data being processed. Such settings can be dynamically expressed
using a closure. For example, we can specify the memory required
as a multiple of the number of cpus. Similarly, we can publish
results to a subfolder based on the sample name.
process FASTQC {
input:
tuple val(sample), path(reads)
script:
"""
fastqc -t $task.cpus $reads
"""
}
// nextflow.config
process {
withName: FASTQC {
cpus = 2
memory = { 2.GB * task.cpus }
publishDir = { "fastqc/$sample" }
}
}
Configuring execution platforms
Nextflow supports a wide range of execution platforms, from running locally, to running on HPC clusters or cloud infrastructures. See https://www.nextflow.io/docs/latest/executor.html for the full list of supported executors.

The default executor configuration is defined within the executor
scope (https://www.nextflow.io/docs/latest/config.html#scope-executor).
For example, in the config below we specify the executor as
Sun Grid Engine, sge and the number of tasks the executor will
handle in a parallel manner (queueSize) to 10.
// nextflow.config
executor {
name = 'sge'
queueSize = 10
}
The process.executor directive allows you to override
the executor to be used by a specific process. This can be
useful, for example, when there are short running tasks
that can be run locally, and are unsuitable for submission
to HPC executors (check for guidelines on best practice use
of your execution system). Other process directives such as
process.clusterOptions, process.queue, and process.machineType
can be also be used to further configure processes depending
on the executor used.
//nextflow.config
executor {
name = 'sge'
queueSize = 10
}
process {
withLabel: 'short' {
executor = 'local'
}
}
Configuring software requirements
An important feature of Nextflow is the ability to manage
software using different technologies. It supports the Conda package
management system, and container engines such as Docker, Singularity,
Podman, Charliecloud, and Shifter. These technologies
allow one to package tools and their dependencies into a software environment
such that the tools will always work as long as the environment can be loaded.
This facilitates portable and reproducible workflows.
Software environment specification is managed from the process scope,
allowing the use of process selectors to manage which processes
load which software environment. Each technology also has its own
scope to provide further technology specific configuration settings.
Software configuration using Conda
Conda is a software package and environment management system that runs on Linux, Windows, and Mac OS. Software packages are bundled into Conda environments along with their dependencies for a particular operating system (Not all software is supported on all operating systems). Software packages are tied to conda channels, for example, bioinformatic software packages are found and installed from the BioConda channel.
A Conda environment can be configured in several ways:
- Provide a path to an existing Conda environment.
- Provide a path to a Conda environment specification file (written in YAML).
- Specify the software package(s) using the
<channel>::<package_name>=<version>syntax (separated by spaces), which then builds the Conda environment when the process is run.
process {
conda = "/home/user/miniconda3/envs/my_conda_env"
withName: FASTQC {
conda = "environment.yml"
}
withName: SALMON {
conda = "bioconda::salmon=1.5.2"
}
}
There is an optional conda scope which allows you to control the
creation of a Conda environment by the Conda package manager.
For example, conda.cacheDir specifies the path where the Conda
environments are stored. By default this is in conda folder of the work directory.
Define a software requirement in the configuration file using conda
Create a config file for the Nextflow script
configuration_fastp.nf. Add a conda directive for the process nameFASTPthat includes the bioconda packagefastp, version 0.12.4-0. Hint You can specify the conda packages using the syntax<channel>::<package_name>=<version>e.g.bioconda::salmon=1.5.2Run the Nextflow scriptconfigure_fastp.nfwith the configuration file using the-coption.// configuration_fastp.nf nextflow.enable.dsl = 2 params.input = "data/yeast/reads/ref1_1.fq.gz" workflow { FASTP( Channel.fromPath( params.input ) ).view() } process FASTP { input: path read output: stdout script: """ fastp -A -i ${read} -o out.fq 2>&1 """ }Solution
// fastp.config process { withName: 'FASTP' { conda = "bioconda::fastp=0.12.4-0" } }nextflow run configure_fastp.nf -c fastp.configN E X T F L O W ~ version 21.04.0 Launching `configuration_fastp.nf` [berserk_jepsen] - revision: 28fadd2486 executor > local (1) [c1/c207d5] process > FASTP (1) [100%] 1 of 1 ✔ Creating Conda env: bioconda::fastp=0.12.4-0 [cache /home/training/work/conda/env-a7a3a0d820eb46bc41ebf4f72d955e5f] ref1_1.fq.gz 58708 Read1 before filtering: total reads: 14677 total bases: 1482377 Q20 bases: 1466210(98.9094%) Q30 bases: 1415997(95.5221%) Read1 after filtering: total reads: 14671 total bases: 1481771 Q20 bases: 1465900(98.9289%) Q30 bases: 1415769(95.5457%) Filtering result: reads passed filter: 14671 reads failed due to low quality: 6 reads failed due to too many N: 0 reads failed due to too short: 0 JSON report: fastp.json HTML report: fastp.html
Software configuration using Docker
Docker is a container technology. Container images are lightweight, standalone, executable package of software that includes everything needed to run an application: code, runtime, system tools, system libraries and settings. Containerized software is intended to run the same regardless of the underlying infrastructure, unlike other package management technologies which are operating system dependant (See the published article on Nextflow). For each container image used, Nextflow uses Docker to spawn an independent and isolated container instance for each process task.
To use Docker, we must provide a container image path using the
process.container directive, and also enable docker in the docker
scope, docker.enabled = true. A container image path takes the form
(protocol://)registry/repository/image:version--build.
By default, Docker containers
run software using a privileged user. This can cause issues,
and so it is also a good idea to supply your user and group
via the docker.runOptions.
process.container = 'quay.io/biocontainers/salmon:1.5.2--h84f40af_0'
docker.enabled = true
docker.runOptions = '-u $(id -u):$(id -g)'
Software configuration using Singularity
Singularity is another container technology, commonly used on
HPC clusters. It is different to Docker in several ways. The
primary differences are that processes are run as the user,
and certain directories are automatically “mounted” (made available)
in the container instance. Singularity also supports building
Singularity images from Docker images, allowing Docker image paths
to be used as values for process.container.
Singularity is enabled in a similar manner to Docker.
A container image path must be provided using process.container and
singularity enabled using singularity.enabled = true.
process.container = 'https://depot.galaxyproject.org/singularity/salmon:1.5.2--h84f40af_0'
singularity.enabled = true
Container protocols
The following protocols are supported:
docker://`: download the container image from the Docker Hub and convert it to the Singularity format (default).library://`: download the container image from the Singularity Library service.shub://`: download the container image from the Singularity Hub.docker-daemon://: pull the container image from a local Docker installation and convert it to a Singularity image file.https://: download the singularity image from the given URL.file://: use a singularity image on local computer storage.
Configuration profiles
One of the most powerful features of Nextflow configuration is to
predefine multiple configurations or profiles for different
execution platforms. This allows a group of predefined settings to
be called with a short invocation, -profile <profile name>.
Configuration profiles are defined in the profiles scope,
which group the attributes that belong to the same profile
using a common prefix.
//configuration_profiles.config
profiles {
standard {
params.genome = '/local/path/ref.fasta'
process.executor = 'local'
}
cluster {
params.genome = '/data/stared/ref.fasta'
process.executor = 'sge'
process.queue = 'long'
process.memory = '10GB'
process.conda = '/some/path/env.yml'
}
cloud {
params.genome = '/data/stared/ref.fasta'
process.executor = 'awsbatch'
process.container = 'cbcrg/imagex'
docker.enabled = true
}
}
This configuration defines three different profiles: standard,
cluster and cloud that set different process configuration
strategies depending on the target execution platform. By
convention the standard profile is implicitly used when no
other profile is specified by the user. To enable a specific
profile use -profile option followed by the profile name:
nextflow run <your script> -profile cluster
Configuration order
Settings from profiles will override general settings in the configuration file. However, it is also important to remember that configuration is evaluated in the order it is read in. For example, in the following example, the
publishDirdirective will always take the value ‘results’ even when the profilehpcis used. This is because the setting is evaluated before Nextflow knows about thehpcprofile. If thepublishDirdirective is moved to after theprofilesscope, thenpublishDirwill use the correct value ofparams.results.params.results = 'results' process.publishDir = params.results profiles { hpc { params.results = '/long/term/storage/results' } }
Inspecting the Nextflow configuration
You can use the command nextflow config to print the resolved
configuration of a workflow. This allows you to see what settings
Nextflow will use to run a workflow.
$ nextflow config workflow_02.nf -profile test
FIXME: fill in
Key Points
Nextflow configuration can be managed using a Nextflow configuration file.
Nextflow configuration files are plain text files containing a set of properties.
You can define process specific settings, such as cpus and memory, within the
processscope.You can assign different resources to different processes using the process selectors
withNameorwithLabel.You can define a profile for different configurations using the
profilesscope. These profiles can be selected when launching a pipeline execution by using the-profilecommand-line optionNextflow configuration settings are evaluated in the order they are read-in.
Workflow configuration settings can be inspected using
nextflow config <script> [options].
Simple RNA-Seq pipeline
Overview
Teaching: 20 min
Exercises: 40 minQuestions
How can I create a Nextflow pipeline from a series of unix commands and input data?
How do I log my pipelines parameters?
How can I manage my pipeline software requirement?
How do I know when my pipeline has finished?
How do I see how much resources my pipeline has used?
Objectives
Create a simple RNA-Seq pipeline.
Use the
log.infofunction to print all the pipeline parameters.Print a confirmation message when the pipeline completes.
Use a conda
environment.ymlfile to install the pipeline’s software requirement.Produce an execution report and generates run metrics from a pipeline run.
We are finally ready to implement a simple RNA-Seq pipeline in Nextflow. This pipeline will have 4 processes that:
- Indexes a transcriptome file.
$ salmon index --threads $task.cpus -t $transcriptome -i index
- Performs quality controls
$ mkdir fastqc_<sample_id>_logs
$ fastqc -o fastqc_<sample_id>_logs -f fastq -q <reads>
- Performs transcript level quantification.
$ salmon quant --threads <cpus> --libType=U -i <index> -1 <read1> -2 <read2> -o <pair_id>
- Create a MultiqQC report form the FastQC and salmon results.
$ multiqc .
To start move to scripts/rnaseq_pipeline folder.
$ cd scripts/rnaseq_pipeline
This folder contains files we will be modifying in this episode.
We will also create a symbolic link to the data directory.
$ ln -s ../../data data
Define the pipeline parameters
The first thing we want to do when writing a pipeline is define the pipeline parameters.
The script script1.nf defines the pipeline input parameters.
//script1.nf
params.reads = "data/yeast/reads/*_{1,2}.fq.gz"
params.transcriptome = "data/yeast/transcriptome/*.fa.gz"
println "reads: $params.reads"
Run it by using the following command:
$ nextflow run script1.nf
We can specify a different input parameter using the --<params> option, for example :
$ nextflow run script1.nf --reads "data/yeast/reads/ref1*_{1,2}.fq.gz"
reads: data/yeast/reads/ref1*_{1,2}.fq.gz
Add a parameter
Modify the
script1.nfadding a third parameter namedoutdirand set it toresults. This parameter will be used as the pipeline output directory.Solution
params.outdir = "results"
It can be useful to print the pipeline parameters to the screen. This can be done using the the log.info command and a multiline string statement. The string method .stripIndent() command is used to remove the indentation on multi-line strings. log.info also saves the output to the log execution file .nextflow.log.
log.info """\
transcriptome: ${params.transcriptome}
"""
.stripIndent()
log.info
Modify the
script1.nfto print all the pipeline parameters by using a singlelog.infocommand and a multiline string statement. See an example here.$ nextflow run script1.nfLook at the output log
.nextflow.log.Solution
Below is an example log.info command printing all the pipeline parameters.
log.info """\ R N A S E Q - N F P I P E L I N E =================================== transcriptome: ${params.transcriptome} reads : ${params.reads} outdir : ${params.outdir} """ .stripIndent()$ less .nextflow.log
Recap
In this step you have learned:
-
How to define parameters in your pipeline script.
-
How to pass parameters by using the command line.
-
The use of
$varand${var}variable placeholders. -
How to use multiline strings.
-
How to use
log.infoto print information and save it in the log execution file.
Create a transcriptome index file
Nextflow allows the execution of any command or user script by using a process definition.
For example,
$ salmon index --threads $task.cpus -t $transcriptome -i index
A process is defined by providing three main declarations:
The second example, script2.nf adds,
- The process
INDEXwhich generate a directory with the index of the transcriptome. This process takes one input, a transcriptome file, and emits one output a salmon index directory. - A queue Channel
transcriptome_chtaking the transcriptome file defined in params variableparams.transcriptome. - Finally the script adds a
workflowdefinition block which calls theINDEXprocess using the Channeltranscriptome_chas input.
//script2.nf
nextflow.enable.dsl=2
/*
* pipeline input parameters
*/
params.reads = "data/yeast/reads/*_{1,2}.fq.gz"
params.transcriptome = "data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
params.outdir = "results"
println """\
R N A S E Q - N F P I P E L I N E
===================================
transcriptome: ${params.transcriptome}
reads : ${params.reads}
outdir : ${params.outdir}
"""
.stripIndent()
/*
* define the `INDEX` process that create a binary index
* given the transcriptome file
*/
process INDEX {
input:
path transcriptome
output:
path 'index'
script:
"""
salmon index --threads $task.cpus -t $transcriptome -i index
"""
}
transcriptome_ch = channel.fromPath(params.transcriptome)
workflow {
INDEX(transcriptome_ch)
}
Try to run it by using the command:
$ nextflow run script2.nf
The execution will fail because the program salmon is not avialable in your environment.
Add the command line option -profile conda to launch the execution through a conda environment as shown below:
$ nextflow run script2.nf -profile conda
This time it works because it uses the conda environment /home/training/miniconda3/envs/nf defined in the nextflow.config` file.
//nextflow.config
profiles {
conda {
process.conda = '/home/training/miniconda3/envs/nf'
}
}
Enable conda by default
Enable the conda execution by removing the profile block in the
nextflow.configfile.Solution
//nextflow.config file process.conda = '/home/training/miniconda3/envs/nf'
View the contents of the index_ch
- Assign the output of the
INDEXprocess to the variableindex_ch.- View the contents of the
index_chchannel by using theviewoperator.Solution
[..truncated..] workflow { index_ch=INDEX(transcriptome_ch) index_ch.view() }
Recap
In this step you have learned:
-
How to define a process executing a custom command
-
How process inputs are declared
-
How process outputs are declared
-
How to use a nextflow configuration file to define and enable a
condaenvironment. -
How to print the content of a channel
view()
Collect read files by pairs
This step shows how to match read files into pairs, so they can be mapped by salmon.
The script script3.nf adds a line to create a channel, read_pairs_ch, containing fastq read pair files using the fromFilePairs channel factory.
//script3.nf
nextflow.enable.dsl = 2
/*
* pipeline input parameters
*/
params.reads = "data/yeast/reads/ref1_{1,2}.fq.gz"
params.transcriptome = "data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
params.outdir = "results"
log.info """\
R N A S E Q - N F P I P E L I N E
===================================
transcriptome: ${params.transcriptome}
reads : ${params.reads}
outdir : ${params.outdir}
"""
.stripIndent()
read_pairs_ch = Channel.fromFilePairs( params.reads )
We can view the contents of the read_pairs_ch by adding the following statement as the last line:
read_pairs_ch.view()
Now if we execute it with the following command:
$ nextflow run script3.nf
It will print an output similar to the one shown below that shows how the read_pairs_ch channel emits a tuple. The tuple is composed of two elements, where the first is the pattern matched by the glob pattern data/yeast/reads/ref1_{1,2}.fq.g, defined by the variable params.reads , and the second is a list representing the actual files.
[..truncated..]
[ref1, [data/yeast/reads/ref1_1.fq.gz,data/yeast/reads/ref1_2.fq.gz]]
To read in other read pairs we can specify a different glob pattern in the params.reads variable by using --reads options on the command line. For example, the following command would read in add the ref samples:
$ nextflow run script3.nf --reads 'data/yeast/reads/ref*_{1,2}.fq.gz'
[..truncated..]
[ref2, [data/yeast/reads/ref2_1.fq.gz, data/yeast/reads/ref2_2.fq.gz]]
[ref3, [data/yeast/reads/ref3_1.fq.gz, data/yeast/reads/ref3_2.fq.gz]]
[ref1, [data/yeast/reads/ref1_1.fq.gz, data/yeast/reads/ref1_2.fq.gz]]
Note File paths including one or more wildcards ie. *, ?, etc. MUST be wrapped in single-quoted characters to avoid Bash expanding the glob pattern on the command line.
We can also add a argument, checkIfExists: true , to the fromFilePairs channel factory to return an message if the file doesn’t exist.
//script3.nf
[..truncated..]
read_pairs_ch = Channel.fromFilePairs( params.reads, checkIfExists: true )
If we now run the script with the --reads parameter data/yeast/reads/*_1,2}.fq.gz
$ nextflow run script3.nf --reads 'data/yeast/reads/*_1,2}.fq.gz'
it will return the message .
[..truncated..]
No such file: data/yeast/reads/*_1,2}.fq.gz
Read in all read pairs
- Add the
checkIfExists: trueargument to thefromFilePairschannel factory inscript3.nf.- Using the command line parameter
--reads, add a glob pattern to read in all the read pairs files from thedata/yeast/readsdirectory.Solution
read_pairs_ch =Channel.fromFilePairs(params.reads, checkIfExists: true)nextflow run script3.nf --reads 'data/yeast/reads/*_{1,2}.fq.gz'[..truncated..] [temp33_1, [data/yeast/reads/temp33_1_1.fq.gz, data/yeast/reads/temp33_1_2.fq.gz]] [ref2, [data/yeast/reads/ref2_1.fq.gz, data/yeast/reads/ref2_2.fq.gz]] [temp33_3, [data/yeast/reads/temp33_3_1.fq.gz, data/yeast/reads/temp33_3_2.fq.gz]] [ref3, [data/yeast/reads/ref3_1.fq.gz, data/yeast/reads/ref3_2.fq.gz]] [temp33_2, [data/yeast/reads/temp33_2_1.fq.gz,data/yeast/reads/temp33_2_2.fq.gz]] [etoh60_2, [data/yeast/reads/etoh60_2_1.fq.gz,data/yeast/reads/etoh60_2_2.fq.gz]] [ref1, [data/yeast/reads/ref1_1.fq.gz, data/yeast/reads/ref1_2.fq.gz]] [etoh60_3, [data/yeast/reads/etoh60_3_1.fq.gz, data/yeast/reads/etoh60_3_2.fq.gz]] [etoh60_1, [data/yeast/reads/etoh60_1_1.fq.gz, data/yeast/reads/etoh60_1_2.fq.gz]]
Recap
In this step you have learned:
-
How to use
fromFilePairsto handle read pair files -
How to use the
checkIfExistsoption to check input file existence
Perform expression quantification
The script script4.nf;
- Adds the quantification process,
QUANT. - Calls the
QUANTprocess in the workflow block.
//script4.nf
..truncated..
/*
* Run Salmon to perform the quantification of expression using
* the index and the matched read files
*/
process QUANT {
input:
path index
tuple val(pair_id), path(reads)
output:
path(pair_id)
script:
"""
salmon quant --threads $task.cpus --libType=U -i $index -1 ${reads[0]} -2 ${reads[1]} -o $pair_id
"""
}
..truncated..
workflow {
read_pairs_ch = Channel.fromFilePairs( params.reads, checkIfExists:true )
transcriptome_ch = Channel.fromPath( params.transcriptome, checkIfExists:true )
index_ch=INDEX(transcriptome_ch)
quant_ch=QUANT(index_ch,read_pairs_ch)
}
The index_ch channel, declared as output in the INDEX process, is used as the first input argument to the QUANT process.
The second input argument of the QUANT process, the read_pairs_ch channel, is a tuple composed of two elements: the pair_id and the reads.
Execute it by using the following command:
$ nextflow run script4.nf
You will see the execution of the index and quantification process.
Re run the command using the -resume option
$ nextflow run script4.nf -resume
The -resume option cause the execution of any step that has been already processed to be skipped.
Try to execute it with more read files as shown below:
$ nextflow run script4.nf -resume --reads 'data/yeast/reads/ref*_{1,2}.fq.gz'
N E X T F L O W ~ version 21.04.0
Launching `script4.nf` [shrivelled_brenner] - revision: c21df6839e
R N A S E Q - N F P I P E L I N E
===================================
transcriptome: data/yeast/transcriptome/Saccharomyces_c
erevisiae.R64-1-1.cdna.all.fa.gz
reads : data/yeast/reads/ref*_{1,2}.fq.gz
outdir : results
executor > local (8)
[02/3742cf] process > INDEX [100%] 1 of 1, cached: 1 ✔
[9a/be3483] process > QUANT (9) [100%] 3 of 3, cached: 1 ✔
You will notice that the INDEX step and one of the QUANT steps has been cached, and
the quantification process is executed more than one time.
When your input channel contains multiple data items Nextflow, where possible, parallelises the execution of your pipeline.
In these situations it is useful to add a tag directive to add some descriptive text to instance of the process being run.
Add a tag directive
Add a
tagdirective to theQUANTprocess ofscript4.nfto provide a more readable execution log.Solution
tag "quantification on $pair_id"
Data produced by the workflow during a process will be saved in the working directory, by default a directory named work.
The working directory should be considered a temporary storage space and any data you wish to save at the end of the workflow should be specified in the process output with the final storage location defined in the publishDir directive.
Note: by default the publishDir directive creates a symbolic link to the files in the working this behaviour can be changed using the mode parameter.
Add a publishDir directive
Add a
publishDirdirective to the quantification process ofscript4.nfto store the process results into folder specified by theparams.outdirNextflow variable. Include thepublishDirmodeoption to copy the output.Solution
publishDir "${params.outdir}/quant", mode:'copy'
Recap
In this step you have learned:
-
How to connect two processes by using the channel declarations.
-
How to resume the script execution skipping already already computed steps.
-
How to use the
tagdirective to provide a more readable execution output. -
How to use the
publishDirto store a process results in a path of your choice.
Quality control
This step implements a quality control step for your input reads. The input to the FASTQC process is the same read_pairs_ch that is provided as input to the quantification process QUANT .
//script5.nf
[..truncated..]
/*
* Run fastQC to check quality of reads files
*/
process FASTQC {
tag "FASTQC on $sample_id"
cpus 1
input:
tuple val(sample_id), path(reads)
output:
path("fastqc_${sample_id}_logs")
script:
"""
mkdir fastqc_${sample_id}_logs
fastqc -o fastqc_${sample_id}_logs -f fastq -q ${reads} -t ${task.cpus}
"""
}
[..truncated..]
workflow {
read_pairs_ch = Channel.fromFilePairs( params.reads, checkIfExists:true )
transcriptome_ch = Channel.fromPath( params.transcriptome, checkIfExists:true )
index_ch=INDEX(transcriptome_ch)
quant_ch=QUANT(index_ch,read_pairs_ch)
}
Run the script script5.nf by using the following command:
$ nextflow run script5.nf -resume
The FASTQC process will not run as the process has not been declared in the workflow scope.
Add FASTQC process
Add the
FASTQCprocess to theworkflow scopeofscript5.nfadding theread_pairs_chchannel as an input. Run the nextflow script using the-resumeoption.$ nextflow run script5.nf -resumeSolution
workflow { read_pairs_ch = Channel.fromFilePairs( params.reads, checkIfExists:true ) transcriptome_ch = Channel.fromPath( params.transcriptome, checkIfExists:true ) index_ch = INDEX( transcriptome_ch ) quant_ch=QUANT(index_ch,read_pairs_ch) fastqc_ch=FASTQC(read_pairs_ch) }
Recap
In this step you have learned:
- How to use the add a
processto theworkflowscope. - Add a channel as input to a
process.
MultiQC report
This step collect the outputs from the quantification and fastqc steps to create a final report by using the MultiQC tool.
The input for the MULTIQC process requires all data in a single channel element.
Therefore, we will need to combine the FASTQC and QUANT outputs using:
- The combining operator
mix: combines the items in the two channels into a single channel
//example of the mix operator
ch1 = Channel.of(1,2)
ch2 = Channel.of('a')
ch1.mix(ch2).view()
1
2
a
- The transformation operator
collectcollects all the items in the new combined channel into a single item.
//example of the collect operator
ch1 = Channel.of(1,2,3)
ch1.collect().view()
[1, 2, 3]
Combing operators
Which is the correct way to combined
mixandcollectoperators so that you have a single channel with one List item?
quant_ch.mix(fastqc_ch).collect()quant_ch.collect(fastqc_ch).mix()fastqc_ch.mix(quant_ch).collect()fastqc_ch.collect(quant_ch).mix()Solution
You need to use the
mixoperator first to combine the channels followed by thecollectoperator to collect all the items in a single item.
In script6.nf we use the statement quant_ch.mix(fastqc_ch).collect() to combine and collect the outputs of the QUANT and FASTQC process to
create the required input for the MULTIQC process.
[..truncated..]
//script6.nf
/*
* Create a report using multiQC for the quantification
* and fastqc processes
*/
process MULTIQC {
publishDir "${params.outdir}/multiqc", mode:'copy'
input:
path('*')
output:
path('multiqc_report.html')
script:
"""
multiqc .
"""
}
workflow {
read_pairs_ch = Channel.fromFilePairs( params.reads, checkIfExists:true )
transcriptome_ch = Channel.fromPath( params.transcriptome, checkIfExists:true )
index_ch=INDEX(transcriptome_ch)
quant_ch=QUANT(index_ch,read_pairs_ch)
fastqc_ch=FASTQC(read_pairs_ch)
MULTIQC(quant_ch.mix(fastqc_ch).collect())
}
Execute the script with the following command:
$ nextflow run script6.nf --reads 'data/yeast/reads/*_{1,2}.fq.gz' -resume
N E X T F L O W ~ version 21.04.0
Launching `script6.nf` [small_franklin] - revision: 9062818659
R N A S E Q - N F P I P E L I N E
===================================
transcriptome: data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz
reads : data/yeast/reads/*_{1,2}.fq.gz
outdir : results
executor > local (9)
[02/3742cf] process > INDEX [100%] 1 of 1, cached: 1 ✔
[9a/be3483] process > QUANT (quantification on etoh60_1) [100%] 9 of 9, cached: 9 ✔
[1f/b7b30a] process > FASTQC (FASTQC on etoh60_1) [100%] 9 of 9, cached: 1 ✔
[2c/206fef] process > MULTIQC [100%] 1 of 1 ✔
It creates the final report in the results folder in the ${params.outdir}/multiqc directory.
Recap
In this step you have learned:
-
How to collect many outputs to a single input with the
collectoperator -
How to mix two channels in a single channel using the
mixoperator. -
How to chain two or more operators togethers using the
.operator.
Handle completion event
This step shows how to execute an action when the pipeline completes the execution.
Note: that Nextflow processes define the execution of asynchronous tasks i.e. they are not executed one after another as they are written in the pipeline script as it would happen in a common imperative programming language.
The script script7..nf uses the workflow.onComplete event handler to print a confirmation message when the script completes.
workflow.onComplete {
log.info ( workflow.success ? "\nDone! Open the following report in your browser --> $params.outdir/multiqc/multiqc_report.html\n" : "Oops .. something went wrong" )
}
This code uses the ternary operator that is a shortcut expression that is equivalent to an if/else branch assigning some value to a variable.
If expression is true? "set value to a" : "else set value to b"
Try to run it by using the following command:
$ nextflow run script7.nf -resume --reads 'data/yeast/reads/*_{1,2}.fq.gz'
[..truncated..]
Done! Open the following report in your browser --> results/multiqc/multiqc_report.html
Metrics and reports
Nextflow is able to produce multiple reports and charts providing several runtime metrics and execution information.
-
The
-with-reportoption enables the creation of the workflow execution report. -
The
-with-traceoption enables the create of a tab separated file containing runtime information for each executed task, including: submission time, start time, completion time, cpu and memory used.. -
The
-with-timelineoption enables the creation of the workflow timeline report showing how processes where executed along time. This may be useful to identify most time consuming tasks and bottlenecks. See an example at this link. -
The
-with-dagoption enables to rendering of the workflow execution direct acyclic graph representation. Note: this feature requires the installation of Graphviz, an open source graph visualization software, in your system.
More information can be found here.
Metrics and reports
Run the script7.nf with the reporting options as shown below:
$ nextflow run script7.nf -resume -with-report -with-trace -with-timeline -with-dag dag.png
- Open the file
report.htmlwith a browser to see the report created with the above command.- Check the content of the file
trace.txtor viewtimeline.htmlto find the longest running process.- View the dag.png
Solution
The
INDEXprocess should be the longest running process. dag.pngThe vertices in the graph represent the pipeline’s processes and operators, while the edges represent the data connections (i.e. channels) between them.
short running tasks
Note: runtime metrics may be incomplete for run short running tasks..
Key Points
Nextflow can combined tasks (processes) and manage data flows using channels into a single pipeline/workflow.
A Workflow can be parameterise using
params. These value of the parameters can be captured in a log file usinglog.infoNextflow can handle a workflow’s software requirements using several technologies including the
condapackage and enviroment manager.Workflow steps are connected via their
inputsandoutputsusingChannels.Intermediate pipeline results can be transformed using Channel
operatorssuch ascombine.Nextflow can execute an action when the pipeline completes the execution using the
workflow.onCompleteevent handler to print a confirmation message.Nextflow is able to produce multiple reports and charts providing several runtime metrics and execution information using the command line options
-with-report,-with-trace,-with-timelineand produce a graph using-with-dag.
Modules
Overview
Teaching: 30 min
Exercises: 15 minQuestions
How can I reuse a Nextflow
processin different workflows?How do I use parameters in a module?
Objectives
Add modules to a Nextflow script.
Create a Nextflow modules.
Understand how to use parameters in a module.
Modules
In most programming languages there is the concept of creating code blocks/modules that can be reused.
Nextflow (DSL2) allows the definition of module scripts that can be included and shared across workflow pipelines.
A module file is nothing more than a Nextflow script containing one or more process definitions that can be imported from another Nextflow script.
A module can contain the definition of a function, process and workflow definitions.
For example:
process INDEX {
input:
path transcriptome
output:
path 'index'
script:
"""
salmon index --threads $task.cpus -t $transcriptome -i index
"""
}
The Nextflow process INDEX above could be saved in a file modules/rnaseq-tasks.nf as a Module script.
Importing module components
A component defined in a module script can be imported into another Nextflow script using the include statement.
For example:
nextflow.enable.dsl=2
include { INDEX } from './modules/rnaseq-tasks'
workflow {
transcriptome_ch = channel.fromPath('data/yeast/transcriptome/*.fa.gz')
//
INDEX(transcriptome_ch)
}
The above snippets includes a process with name INDEX defined in the module script rnaseq-tasks.nf in the main execution context, as such it can be invoked in the workflow scope.
Nextflow implicitly looks for the script file ./modules/rnaseq-tasks.nf resolving the path against the including script location.
Note: Relative paths must begin with the ./ prefix.
Remote
You can not include a script from a remote URL in the
fromstatement.
Add module
Add the Nextflow module
FASTQCfrom the Nextflow script./modules/rnaseq-tasks.nfto the following workflow.nextflow.enable.dsl=2 params.reads = "data/yeast/reads/ref1_{1,2}.fq.gz" read_pairs_ch = channel.fromFilePairs( params.reads, checkIfExists:true ) workflow { FASTQC(read_pairs_ch) }Solution
nextflow.enable.dsl=2 include { FASTQC } from './modules/rnaseq-tasks' params.reads = "$baseDir/data/yeast/reads/ref1_{1,2}.fq.gz" read_pairs_ch = channel.fromFilePairs( params.reads, checkIfExists:true ) workflow { FASTQC(read_pairs_ch) }
Multiple inclusions
A Nextflow script allows the inclusion of any number of modules.
When multiple components need to be included from the some module script,
the component names can be specified in the same inclusion using the curly brackets {}.
Note Component names are separated by a semi-colon ; as shown below:
nextflow.enable.dsl=2
include { INDEX; QUANT } from './modules/rnaseq-tasks'
workflow {
reads = channel.fromFilePairs('data/yeast/reads/*_{1,2}.fq.gz')
transcriptome_ch = channel.fromPath('data/yeast/transcriptome/*.fa.gz')
INDEX(transcriptome_ch)
QUANT(index.out,reads)
}
Module aliases
A process component, such as INDEX, can be invoked only once in the same workflow context.
However, when including a module component it’s possible to specify a name alias using the keyword as in the include statement. This allows the inclusion and the invocation of the same component multiple times in your script using different names.
For example:
nextflow.enable.dsl=2
include { INDEX } from './modules/rnaseq-tasks'
include { INDEX as SALMON_INDEX } from './modules/rnaseq-tasks'
workflow {
transcriptome_ch = channel.fromPath('data/yeast/transcriptome/*.fa.gz')
INDEX(transcriptome_ch)
SALMON_INDEX(transcriptome_ch)
}
In the above script the INDEX process is imported as INDEX and an alias SALMON_INDEX.
The same is possible when including multiple components from the same module script as shown below:
nextflow.enable.dsl=2
include { INDEX; INDEX as SALMON_INDEX } from './modules/rnaseq-tasks'
workflow {
transcriptome_ch = channel.fromPath('/data/yeast/transcriptome/*.fa.gz)'
INDEX(transcriptome)
SALMON_INDEX(transcriptome)
}
Add multiple modules
Add the Nextflow modules
FASTQCandMULTIQCfrom the Nextflow scriptmodules/rnaseq-tasks.nfto the following workflow.nextflow.enable.dsl=2 params.reads = "$baseDir/data/yeast/reads/ref1_{1,2}.fq.gz" read_pairs_ch = channel.fromFilePairs( params.reads, checkIfExists:true ) workflow { FASTQC(read_pairs_ch) MULTIQC(fastqc.out.collect()) }Solution
nextflow.enable.dsl=2 include { FASTQC; MULTIQC } from './modules/rnaseq-tasks' params.reads = "$baseDir/data/yeast/reads/ref1_{1,2}.fq.gz" read_pairs_ch = channel.fromFilePairs( params.reads, checkIfExists:true ) workflow { FASTQC(read_pairs_ch) MULTIQC(fastqc.out.collect()) }
Module parameters
A module script can define one or more parameters using the same syntax of a Nextflow workflow script:
//functions.nf file
params.message = 'parameter from module script'
//The def keyword allows use to define a function that we can use in the code
def sayMessage() {
println "$params.message"
}
Then, parameters are inherited from the including context. For example:
nextflow.enable.dsl=2
params.message = 'parameter from workflow script'
include {sayMessage} from './modules/functions'
workflow {
sayMessage()
}
The above snippet prints:
parameter from workflow script
The module uses the parameters define before the include statement, therefore any further parameter set later is ignored.
Tip: Define all pipeline parameters at the beginning of the script before any include declaration.
The option addParams can be used to extend the module parameters without affecting the parameters set before the include statement.
For example:
nextflow.enable.dsl=2
params.message = 'parameter from workflow script'
include {sayMessage} from './modules/module.nf' addParams(message: 'using addParams')
workflow {
sayMessage()
}
The above code snippet prints:
using addParams
Key Points
A module file is a Nextflow script containing one or more
processdefinitions that can be imported from another Nextflow script.To import a module into a workflow use the
includekeyword.A module script can define one or more parameters using the same syntax of a Nextflow workflow script.
The module inherits the parameters define before the include statement, therefore any further parameter set later is ignored.
Sub-workflows
Overview
Teaching: 20 min
Exercises: 0 minQuestions
How do I reuse a workflow as part of a larger workflow?
How do I run only a part of a workflow?
Objectives
Understand how to create a sub-workflow.
Understand how to run part of a workflow.
Sub-workflows
We have seen previously the Nextflow DSL2 syntax allows for the definition of reusable processes (modules). Nextflow DSL2 also allow the definition reusable sub-workflow libraries.
Workflow definition
The workflow keyword allows the definition of workflow components that enclose the invocation of one or more processes and operators.
For example,:
nextflow.enable.dsl=2
include {QUANT;INDEX} from './modules/module.nf'
workflow RNASEQ_QUANT_PIPE {
read_pairs_ch = channel.fromFilePairs('data/yeast/reads/*_{1,2}.fq.gz')
transcriptome_ch = channel.fromPath('/data/yeast/transcriptome/*.fa.gz')
QUANT(INDEX(transcriptome_ch),read_pairs_ch)
}
The above snippet defines a workflow component, named RNASEQ_QUANT_PIPE, that can be invoked from another workflow component definition in the same way as any other function or process i.e. RNASEQ_QUANT().
nextflow.enable.dsl=2
include {QUANT;INDEX} from './modules/module.nf'
workflow RNASEQ_QUANT_PIPE {
read_pairs_ch = channel.fromFilePairs('data/yeast/reads/*_{1,2}.fq.gz')
transcriptome_ch = channel.fromPath('/data/yeast/transcriptome/*.fa.gz')
QUANT(INDEX(transcriptome_ch),read_pairs_ch)
}
// Implicit workflow
workflow {
/*
* Call sub-workflow using <WORKFLOWNAME>() syntax
*/
RNASEQ_QUANT_PIPE()
}
Implicit workflow
A workflow definition which does not declare any name is assumed to be the main workflow, and it is implicitly executed. Therefore it’s the entry point of the workflow application.
Workflow parameters
A workflow component can access any variable and parameter defined in the outer scope.
For Example:
nextflow.enable.dsl=2
include {QUANT;INDEX} from './modules/module.nf'
params.transcriptome = '/some/data/file'
read_pairs_ch = channel.fromFilePairs('data/yeast/reads/*_{1,2}.fq.gz')
workflow RNASEQ_QUANT_PIPE {
transcriptome_ch = channel.fromPath(params.transcriptome)
QUANT(INDEX(transcriptome_ch),read_pairs_ch)
}
Workflow inputs
A workflow component can declare one or more input channels using the take keyword.
For example:
nextflow.enable.dsl=2
include {QUANT;INDEX} from './modules/module.nf'
params.transcriptome = '/some/data/file'
read_pairs_ch = channel.fromFilePairs('data/yeast/reads/*_{1,2}.fq.gz')
workflow RNASEQ_QUANT_PIPE {
take:
transcriptome_ch
read_pairs_ch
main:
transcriptome_ch = channel.fromPath(params.transcriptome)
INDEX(transcriptome_ch)
QUANT(INDEX.out,read_pairs_ch)
}
Warning
When the
takekeyword is used, the beginning of the workflow body needs to be identified with themainkeyword. Then, the input can be specified as an argument in the workflow invocation statement:
These input channels can then be passed to the workflow as parameters inside the (). Multiple parameters are separated by a comma , and must be specified in the order they appear under take:
nextflow.enable.dsl=2
include {QUANT;INDEX} from './modules/module.nf'
params.transcriptome = '/some/data/file'
read_pairs_ch = channel.fromFilePairs('data/yeast/reads/*_{1,2}.fq.gz')
workflow RNASEQ_QUANT_PIPE {
take:
transcriptome_ch
read_pairs_ch
main:
transcriptome_ch = channel.fromPath(params.transcriptome)
INDEX(transcriptome_ch)
QUANT(INDEX.out,read_pairs_ch)
}
workflow {
RNASEQ_QUANT_PIPE(transcriptome_ch,read_pairs_ch )
}
Note
Workflow inputs are by definition channel data structures. If a basic data type is provided instead, ie. number, string, list, etc. it’s implicitly converted to a channel value (ie. non-consumable).
Workflow outputs
A workflow component can declare one or more output channels using the emit keyword.
For example:
nextflow.enable.dsl=2
include {QUANT;INDEX} from './modules/module.nf'
params.transcriptome = '/some/data/file'
read_pairs_ch = channel.fromFilePairs('data/yeast/reads/*_{1,2}.fq.gz')
workflow RNASEQ_QUANT_PIPE {
take:
transcriptome_ch
read_pairs_ch
emit:
QUANT.out
main:
transcriptome_ch = channel.fromPath(params.transcriptome)
INDEX(transcriptome_ch)
QUANT(INDEX.out,read_pairs_ch)
}
The above script declares one output, QUANT.out.
The result of the RNASEQ_QUANT_PIPE execution can be accessed using the out property ie. RNASEQ_QUANT_PIPE.out.
When there are multiple output channels declared, use the array bracket notation to access each output component as described for the Process outputs definition.
RNASEQ_QUANT_PIPE.out[0]
RNASEQ_QUANT_PIPE.out[1]
Alternatively, the output channel can be accessed using a name which it’s assigned to in the emit declaration:
For example:
nextflow.enable.dsl=2
workflow RNASEQ_QUANT_PIPE {
main:
INDEX(transcriptome_ch)
QUANT(INDEX.out,read_pairs_ch)
emit:
read_quant = QUANT.out
}
The output QUANT.out is assigned the name read_quant
The the result of the above snippet can accessed using:
RNASEQ_QUANT_PIPE.out.read_quant`.
Note
Implicit workflow definition is ignored when a script is included as module. This allows the writing a workflow script that can be used either as a library module and as application script.
Workflow composition
As with modules workflows components can be defined within your script or imported by a include statment. After which thet can then be invoked and composed as any other workflow component or process in your script.
nextflow.enable.dsl=2
// file modules/qc.nf
include {FASTQC} from './modules.nf'
workflow READ_QC_PIPE {
take:
read_pairs_ch
quant_out_ch
main:
FASTQC(read_pairs_ch)
emit:
FASTQC.out
}
nextflow.enable.dsl=2
include { READ_QC_PIPE } from './modules/qc.nf'
workflow RNASEQ_QUANT_PIPE {
take:
transcriptome_ch
read_pairs_ch
main:
INDEX(transcriptome)
QUANT(INDEX.out)
emit:
QUANT.out
}
params.transcriptome = '/some/data/file'
read_pairs_ch = channel.fromFilePairs('data/yeast/reads/*_{1,2}.fq.gz')
transcriptome_ch = channel.fromPath(params.transcriptome)
workflow {
take:
transcriptome_ch
read_pairs_ch
main:
RNASEQ_QUANT(transcriptome_ch,read_pairs_ch)
READ_QC(read_pairs_ch,RNASEQ_QUANT.out)
MULTIQC(RNASEQ_QUANT.out.mix(READ_QC).collect())
}
Nested workflow execution
Nested workflow execution determines an implicit scope. Therefore the same process can be invoked in two different workflow scopes, like for example in the above snippet
INDEXcould be used either inRNASEQ_QUANTandRNASEQ_QC. The workflow execution path along with the process names defines the process fully qualified name that is used to distinguish the two different process invocations i.e.RNASEQ_QUANT:INDEXandRNASEQ_QC:INDEXin the above example.
Specific workflow entry points
By default, the unnamed workflow is assumed to be the main entry point for the script. Using named workflows, the entry point can be customised by using the entry option of the run command. This allows users to run a specific sub-workflow or a section of their entire workflow script.
For example:
$ nextflow run main.nf -entry RNASEQ_QUANT_PIPE
The above command would run the RNASEQ_QUANT_PIPE sub-workflow.
Key Points
Nextflow allows for definition of reusable sub-workflow libraries.
Sub-workflow allows the definition of workflow processes that can be included from any other script and invoked as a custom function within the new workflow scope. This enables reuse of workflow components
The
entryoption of the nextflowruncommand specifies the workflow name to be executed
Reporting
Overview
Teaching: 20 min
Exercises: 5 minQuestions
How do I get information about my pipeline run?
How can I see what commands I ran?
How can I create a report from my run?
Objectives
View Nextflow pipeline run logs.
Use
nextflow logto view more information about a specific run.Create an HTML report from a pipeline run.
Nextflow log
Once a script has run, Nextflow stores a log of all the workflows executed in the current folder. Similar to an electronic lab book, this means you have a record of all processing steps and commands run.
You can print Nextflow’s execution history and log information using the nextflow log command.
$ nextflow log
TIMESTAMP DURATION RUN NAME STATUS REVISION ID SESSION ID COMMAND
This will print a summary of the executions log and runtime information for all pipelines run. By default, included in the summary, are the date and time it ran, how long it ran for, the run name, run status, a revision ID, the session id and the command run on the command line.
Show Execution Log
Listing the execution logs of previous invocations of all pipelines in a directory.
$ nextflow logSolution
The output will look similar to this:
TIMESTAMP DURATION RUN NAME STATUS REVISION ID SESSION ID COMMAND 2021-03-19 13:45:53 6.5s fervent_babbage OK c54a707593 15487395-443a-4835-9198-229f6ad7a7fd nextflow run wc.nf 2021-03-19 13:46:53 6.6s soggy_miescher OK c54a707593 58da0ccf-63f9-42e4-ba4b-1c349348ece5 nextflow run wc.nf --samples 'data/yeast/reads/*.fq.gz'
Pipeline execution report
If we want to get more information about an individual run we can add the run name or session ID to the log command.
For example:
$ nextflow log tiny_fermat
/data/.../work/7b/3753ff13b1fa5348d2d9b6f512153a
/data/.../work/c1/56a36d8f498c99ac6cba31e85b3e0c
/data/.../work/f7/659c65ef60582d9713252bcfbcc310
/data/.../work/82/ba67e3175bd9e6479d4310e5a92f99
/data/.../work/e5/2816b9d4e7b402bfdd6597c2c2403d
/data/.../work/3b/3485d00b0115f89e4c202eacf82eba
This will list the work directory for each process.
Task ID
The task ID is a a 32 hexadecimal digit,e.g.
3b3485d00b0115f89e4c202eacf82eba. A task’s unique ID is generated as a 128-bit hash number obtained from a composition of the task’s:
- Inputs values
- Input files
- Command line string
- Container ID
- Conda environment
- Environment modules
- Any executed scripts in the bin directory
Fields
If we want to print more metadata we can use the log command and the option -f (fields) followed by a comma delimited list of fields.
This can be composed to track the provenance of a workflow result.
For example:
$ nextflow log tiny_fermat -f 'process,exit,hash,duration'
Will output the process name, exit status, hash and duration of the process for the tiny_fermat run to the terminal.
index 0 7b/3753ff 2s
fastqc 0 c1/56a36d 9.3s
fastqc 0 f7/659c65 9.1s
quant 0 82/ba67e3 2.7s
quant 0 e5/2816b9 3.2s
multiqc 0 3b/3485d0 6.3s
The complete list of available fields can be retrieved with the command:
$ nextflow log -l
attempt
complete
container
cpus
disk
duration
env
error_action
exit
hash
inv_ctxt
log
memory
module
name
native_id
pcpu
peak_rss
peak_vmem
pmem
process
queue
rchar
read_bytes
realtime
rss
scratch
script
start
status
stderr
stdout
submit
syscr
syscw
tag
task_id
time
vmem
vol_ctxt
wchar
workdir
write_bytes
Script
If we want a log of all the commands executed in the pipeline we can use the script
field. It is important to note that the resultant output can not be used to run the pipeline steps.
Filtering
The output from the log command can be very long. We can subset the output using the option -F (filter) specifying the filtering criteria. This will print only those tasks matching a pattern using the syntax =~/<pattern>/.
For example to filter for process with the name fastqc we would run:
$ nextflow log tiny_fermat -F 'process =~ /fastqc/'
/data/.../work/c1/56a36d8f498c99ac6cba31e85b3e0c
/data/.../work/f7/659c65ef60582d9713252bcfbcc310
This can be useful to locate specific tasks work directories.
View run log
Use the Nextflow
logcommand specifying arun nameand the fields. name, hash, process and statusSolution
Example solution using run name
elegant_descartes.$ nextflow log elegant_descartes -f name,hash,process,statusFilter pipeline run log
Use the
-Foption and a regular expression to filter the for a specific process e.g. multiqc.Solution
$ nextflow log elegant_descartes -f name,hash,process,status -F 'process =~ /multiqc/'
Templates
The -t option allows a template (string or file) to be specified. This makes it possible to create a custom report in any text based format.
For example you could save this markdown snippet to a file e.g. my-template.md:
## $name
script:
$script
exist status: $exit
task status: $status
task folder: $workdir
Then, the following log command will output a markdown file containing the script, exit status and folder of all executed tasks:
$ nextflow log elegant_descartes -t my-template.md > execution-report.md
Or, the template file can also be written in HTML.
For example:
<div>
<h2>${name}</h2>
<div>
Script:
<pre>${script}</pre>
</div>
<ul>
<li>Exit: ${exit}</li>
<li>Status: ${status}</li>
<li>Work dir: ${workdir}</li>
<li>Container: ${container}</li>
</ul>
</div>
By saving the above snippet in a file named template.html, you can run the following command:
$ nextflow log elegant_descartes -t template.html > provenance.html
To view the report open it in a browser.
Generate an HTML run report
Generate an HTML report for a run using the
-toption and the template.html file.Solution
$ nextflow log elegant_descartes -t template.html > provenance.html
Key Points
Nextflow can produce a custom execution report with run information using the
logcommand.You can generate a report using the
-toption specifying a template file.
Workflow caching and checkpointing
Overview
Teaching: 20 min
Exercises: 10 minQuestions
How can I restart a Nextflow workflow after an error?
How can I add new data to a workflow without starting from the beginning?
Where can I find intermediate data and results?
Objectives
Resume a Nextflow workflow using the
-resumeoption.Restart a Nextflow workflow using new data.
A key feature of workflow management systems, like Nextflow, is re-entrancy, which is the ability to restart a pipeline after an error from the last successfully executed process. Re-entrancy enables time consuming successfully completed steps, such as index creation, to be skipped when adding more data to a pipeline. This in turn leads to faster prototyping and development of workflows, and faster analyses of additional data. Nextflow achieves re-entrancy by automatically keeping track of all the processes executed in your pipeline via caching and checkpointing.
Resume
To restart from the last successfully executed process we add the command line option -resume to the Nextflow command.
For example, the command below would resume the wc.nf script from the last successful process.
$ nextflow run wc.nf --input 'data/yeast/reads/ref1*.fq.gz' -resume
We can see in the output that the results from the process NUM_LINES has been retrieved from the cache.
Launching `wc.nf` [condescending_dalembert] - revision: fede04a544
[c9/2597d5] process > NUM_LINES (1) [100%] 2 of 2, cached: 2 ✔
ref1_1.fq.gz 58708
ref1_2.fq.gz 58708
Resume a pipeline
Resume the Nextflow script
wc.nfby re-running the command and adding the parameter-resumeand the parameter--input 'data/yeast/reads/temp33*':Solution
$ nextflow run wc.nf --input 'data/yeast/reads/temp33*' -resumeIf your previous run was successful the output will look similar to this:
N E X T F L O W ~ version 20.10.0 Launching `wc.nf` [nauseous_leavitt] - revision: fede04a544 [21/6116de] process > NUM_LINES (4) [100%] 6 of 6, cached: 6 ✔ temp33_3_2.fq.gz 88956 temp33_3_1.fq.gz 88956 temp33_1_1.fq.gz 82372 temp33_2_2.fq.gz 63116 temp33_1_2.fq.gz 82372 temp33_2_1.fq.gz 63116You will see that the execution of the process
NUMLINESis actually skipped (cached text appears), and the results are retrieved from the cache.
How does resume work?
The mechanism works by assigning a unique ID to each task. This unique ID is used to create a separate execution directory, within the work directory, where the tasks are executed and the results stored. A task’s unique ID is generated as a 128-bit hash number obtained from a composition of the task’s:
- Inputs values
- Input files
- Command line string
- Container ID
- Conda environment
- Environment modules
- Any executed scripts in the bin directory
When we resume a workflow Nextflow uses this unique ID to check if:
- The working directory exists
- It contains a valid command exit status
- It contains the expected output files.
If these conditions are satisfied, the task execution is skipped and the previously computed outputs are applied. When a task requires recomputation, ie. the conditions above are not fulfilled, the downstream tasks are automatically invalidated.
Therefore, if you modify some parts of your script, or alter the input data using -resume, will only execute the processes that are actually changed.
The execution of the processes that are not changed will be skipped and the cached result used instead.
This helps a lot when testing or modifying part of your pipeline without having to re-execute it from scratch.
Modify Nextflow script and re-run.
Alter the timestamp on the file temp33_3_2.fq.gz using the UNIX
touchcommand.$ touch data/yeast/reads/temp33_3_2.fq.gzRun command below.
$ nextflow run wc.nf --input 'data/yeast/reads/temp33*' -resumeHow many processes will be cached and how many will run ?
Solution
The output will look similar to this:
N E X T F L O W ~ version 20.10.0 Launching `wc.nf` [gigantic_minsky] - revision: fede04a544 executor > local (1) [20/cda0d5] process > NUM_LINES (5) [100%] 6 of 6, cached: 5 ✔ temp33_1_2.fq.gz 82372 temp33_3_1.fq.gz 88956 temp33_2_1.fq.gz 63116 temp33_1_1.fq.gz 82372 temp33_2_2.fq.gz 63116 temp33_3_2.fq.gz 88956As you changed the timestamp on one file it will only re-run that process. The results for the other 5 processes are retrieved from the cache.
The Work directory
By default the pipeline results are cached in the directory work where the pipeline is launched.
We can use the Bash tree command to list the contents of the work directory.
Note: By default tree does not print hidden files (those beginning with a dot .). Use the -a to view all files.
$ tree -a work
Example output
work/
├── 12
│ └── 5489f3c7dbd521c0e43f43b4c1f352
│ ├── .command.begin
│ ├── .command.err
│ ├── .command.log
│ ├── .command.out
│ ├── .command.run
│ ├── .command.sh
│ ├── .exitcode
│ └── temp33_1_2.fq.gz -> /home/training/data/yeast/reads/temp33_1_2.fq.gz
├── 3b
│ └── a3fb24ad3242e4cc8e5aa0c24d174b
│ ├── .command.begin
│ ├── .command.err
│ ├── .command.log
│ ├── .command.out
│ ├── .command.run
│ ├── .command.sh
│ ├── .exitcode
│ └── temp33_2_1.fq.gz -> /home/training/data/yeast/reads/temp33_2_1.fq.gz
├── 4c
│ └── 125b5e5a5ee144fa25dd9bccd467e9
│ ├── .command.begin
│ ├── .command.err
│ ├── .command.log
│ ├── .command.out
│ ├── .command.run
│ ├── .command.sh
│ ├── .exitcode
│ └── temp33_3_1.fq.gz -> /home/training/data/yeast/reads/temp33_3_1.fq.gz
├── 54
│ └── eb9d72e9ac24af8183de569ab0b977
│ ├── .command.begin
│ ├── .command.err
│ ├── .command.log
│ ├── .command.out
│ ├── .command.run
│ ├── .command.sh
│ ├── .exitcode
│ └── temp33_2_2.fq.gz -> /home/training/data/yeast/reads/temp33_2_2.fq.gz
├── e9
│ └── 31f28c291481342cc45d4e176a200a
│ ├── .command.begin
│ ├── .command.err
│ ├── .command.log
│ ├── .command.out
│ ├── .command.run
│ ├── .command.sh
│ ├── .exitcode
│ └── temp33_1_1.fq.gz -> /home/training/data/yeast/reads/temp33_1_1.fq.gz
└── fa
└── cd3e49b63eadd6248aa357083763c1
├── .command.begin
├── .command.err
├── .command.log
├── .command.out
├── .command.run
├── .command.sh
├── .exitcode
└── temp33_3_2.fq.gz -> /home/training/data/yeast/reads/temp33_3_2.fq.gz
Task execution directory
Within the work directory there are multiple task execution directories.
There is one directory for each time a process is executed.
These task directories are identified by the process execution hash. For example
the task directory fa/cd3e49b63eadd6248aa357083763c1 would be location for the process identified by the hash fa/cd3e49 .
The task execution directory contains:
-
.command.sh: The command script. -
.command.run: The command wrapped used to run the job. -
.command.out: The complete job standard output. -
.command.err: The complete job standard error. -
.command.log: The wrapper execution output. -
.command.begin: A file created as soon as the job is launched. -
.exitcode: A file containing the task exit code. -
Any task input files (symlinks)
-
Any task output files
Specifying another work directory
Depending on your script, this work folder can take a lot of disk space.
You can specify another work directory using the command line option -w.
Note Using a different work directory will mean that any jobs will need to re-run from the beginning.
$ nextflow run wc.nf --input 'data/yeast/reads/temp33*' -w second_work_dir -resume
N E X T F L O W ~ version 21.04.0
Launching `wc.nf` [deadly_easley] - revision: fede04a544
executor > local (6)
[9d/0f5e89] process > NUM_LINES (5) [100%] 6 of 6 ✔
temp33_3_2.fq.gz 88956
temp33_1_1.fq.gz 82372
temp33_3_1.fq.gz 88956
temp33_1_2.fq.gz 82372
temp33_2_2.fq.gz 63116
temp33_2_1.fq.gz 63116
Clean the work directory
If you are sure you won’t resume your pipeline execution, clean this folder periodically using the command nextflow clean.
$ nextflow clean [run_name|session_id] [options]
Supply the option -n to print names of files to be removed without deleting them, or -f to force the removal of the files. If you only want to remove files from a run but retain execution log entries and metadata, add the option -k. Multiple runs can be cleaned with the options, -before, -after or -but before the run name.
For example, the command below would remove all the temporary files and log entries for runs before the run gigantic_minsky.
$ nextflow clean -f -before gigantic_minsky
Remove a Nextflow run.
Remove the last Nextflow run using the command
nextflow clean. First use the option-dry-runto see which files would be deleted and then re-run removing the run and associated files.Solution
An example nextflow clean command with
dry-run.$ nextflow clean nauseous_leavitt -dry-runAn example nextflow clean command removing the files.
$ nextflow clean nauseous_leavitt -f
Key Points
Nextflow automatically keeps track of all the processes executed in your pipeline via checkpointing.
Nextflow caches intermediate data in task directories within the work directory.
Nextflow caching and checkpointing allows re-entrancy into a workflow after a pipeline error or using new data, skipping steps that have been successfully executed. - Re-entrancy is enabled using the
-resumeoption.
Deploying nf-core pipelines
Overview
Teaching: 30 min
Exercises: 10 minQuestions
Where can I find best-practice Nextflow bioinformatic pipelines?
How do I run nf-core pipelines?
How do I configure nf-core pipelines to use my data?
How do I reference nf-core pipelines?
Objectives
Understand what nf-core is and how it relates to Nextflow.
Use the nf-core helper tool to find nf-core pipelines.
Understand how to configuration nf-core pipelines.
Run a small nf-core pipeline using a test dataset.
What is nf-core?
nf-core is a community-led project to develop a set of best-practice pipelines built using Nextflow workflow management system. Pipelines are governed by a set of guidelines, enforced by community code reviews and automatic code testing.

In this episode we will covering finding, deploying and configuring nf-core pipelines.
What are nf-core pipelines?
nf-core pipelines are an organised collection of Nextflow scripts, other non-nextflow scripts (written in any language), configuration files, software specifications, and documentation hosted on GitHub. There is generally a single pipeline for a given data and analysis type e.g. There is a single pipeline for bulk RNA-Seq. All nf-core pipelines are distributed under the, permissive free software, MIT licences.
What is nf-core tools?
nf-core provides a suite of helper tools aim to help people run and develop pipelines. The nf-core tools package is written in Python and can run from the command line or imported and used within other packages.
Automatic version check
nf-core/tools automatically checks the web to see if there is a new version of nf-core/tools available. If you would prefer to skip this check, set the environment variable NFCORE_NO_VERSION_CHECK. For example:
export NFCORE_NO_VERSION_CHECK=1
nf-core tools sub-commands
You can use the --help option to see the range of nf-core tools sub-commands.
In this episode we will be covering the list, launch and download sub-commands which
aid in the finding and deployment of the nf-core pipelines.
$ nf-core --help
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 2.1
Usage: nf-core [OPTIONS] COMMAND [ARGS]...
Options:
--version Show the version and exit.
-v, --verbose Print verbose output to the console.
-l, --log-file <filename> Save a verbose log to a file.
--help Show this message and exit.
Commands:
list List available nf-core pipelines with local info.
launch Launch a pipeline using a web GUI or command line prompts.
download Download a pipeline, nf-core/configs and pipeline...
licences List software licences for a given workflow.
create Create a new pipeline using the nf-core template.
lint Check pipeline code against nf-core guidelines.
modules Tools to manage Nextflow DSL2 modules as hosted on...
schema Suite of tools for developers to manage pipeline schema.
bump-version Update nf-core pipeline version number.
sync Sync a pipeline TEMPLATE branch with the nf-core template.
Listing available nf-core pipelines
The simplest sub-command is nf-core list, which lists all available nf-core pipelines in the nf-core Github repository.
The output shows the latest version number and when that was released. If the pipeline has been pulled locally using Nextflow, it tells you when that was and whether you have the latest version.
Run the command below.
$ nf-core list
An example of the output from the command is as follows:
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 2.1
┏━━━━━━━━━━━━━━━━━━━┳━━━━━━━┳━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━┓
┃ Pipeline Name ┃ Stars ┃ Latest Release ┃ Released ┃ Last Pulled ┃ Have latest release? ┃
┡━━━━━━━━━━━━━━━━━━━╇━━━━━━━╇━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━┩
│ mhcquant │ 14 │ 2.0.0 │ 4 days ago │ - │ - │
│ bacass │ 28 │ 2.0.0 │ 2 weeks ago │ - │ - │
│ viralrecon │ 48 │ 2.2 │ 1 months ago │ - │ - │
│ rnaseq │ 364 │ 3.3 │ 1 months ago │ - │ - │
│ mag │ 63 │ 2.1.0 │ 1 months ago │ - │ - │
│ fetchngs │ 25 │ 1.2 │ 1 months ago │ - │ - │
│ bcellmagic │ 14 │ 2.0.0 │ 2 months ago │ - │ - │
│ ampliseq │ 67 │ 2.0.0 │ 2 months ago │ - │ - │
[..truncated..]
Filtering available nf-core pipelines
If you supply additional keywords after the list sub-command, the listed pipeline will be filtered.
Note: that this searches more than just the displayed output, including keywords and description text.
Here we filter on the keywords rna and rna-seq .
$ nf-core list rna rna-seq
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 2.1
┏━━━━━━━━━━━━━━━┳━━━━━━━┳━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━┳━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━┓
┃ Pipeline Name ┃ Stars ┃ Latest Release ┃ Released ┃ Last Pulled ┃ Have latest release? ┃
┡━━━━━━━━━━━━━━━╇━━━━━━━╇━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━╇━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━┩
│ rnaseq │ 364 │ 3.3 │ 1 months ago │ - │ - │
│ smrnaseq │ 26 │ 1.1.0 │ 3 months ago │ - │ - │
│ dualrnaseq │ 4 │ 1.0.0 │ 7 months ago │ - │ - │
│ rnafusion │ 60 │ 1.2.0 │ 1 years ago │ - │ - │
│ circrna │ 8 │ dev │ - │ - │ - │
│ lncpipe │ 22 │ dev │ - │ - │ - │
│ scflow │ 6 │ dev │ - │ - │ - │
└───────────────┴───────┴────────────────┴──────────────┴─────────────┴──────────────────────┘
Sorting available nf-core pipelines
You can sort the results by adding the option --sort followed by a keyword.
For example, latest release (--sort release), when you last pulled a local copy (--sort pulled), alphabetically (--sort name), or number of GitHub stars (--sort stars).
nf-core list rna rna-seq --sort stars
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 2.1
┏━━━━━━━━━━━━━━━┳━━━━━━━┳━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━┳━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━┓
┃ Pipeline Name ┃ Stars ┃ Latest Release ┃ Released ┃ Last Pulled ┃ Have latest release? ┃
┡━━━━━━━━━━━━━━━╇━━━━━━━╇━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━╇━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━┩
│ rnaseq │ 364 │ 3.3 │ 1 months ago │ - │ - │
│ rnafusion │ 60 │ 1.2.0 │ 1 years ago │ - │ - │
│ smrnaseq │ 26 │ 1.1.0 │ 3 months ago │ - │ - │
│ lncpipe │ 22 │ dev │ - │ - │ - │
│ circrna │ 8 │ dev │ - │ - │ - │
│ scflow │ 6 │ dev │ - │ - │ - │
│ dualrnaseq │ 4 │ 1.0.0 │ 7 months ago │ - │ - │
└───────────────┴───────┴────────────────┴──────────────┴─────────────┴──────────────────────┘
Archived pipelines
Archived pipelines are not returned by default. To include them, use the
--show_archivedflag.
Exercise: listing nf-core pipelines
- Use the
--helpflag to print the list command usage.- Sort all pipelines by popularity (stars) and find out which is the most popular?.
- Filter pipelines for those that work with RNA and find out how many there are?
Solution
Use the
--helpflag to print the list command usage.$ nf-core list --helpSort all pipelines by popularity (stars).
$ nf-core list --sort starsFilter pipelines for those that work with RNA.
$ nf-core list rna
Running nf-core pipelines
Software requirements for nf-core pipelines
nf-core pipeline software dependencies are specified using either Docker, Singularity or Conda. It is Nextflow that handles the downloading of containers and creation of conda environments. In theory it is possible to run the pipelines with software installed by other methods (e.g. environment modules, or manual installation), but this is not recommended.
Fetching pipeline code
Unless you are actively developing pipeline code, you should use Nextflow’s built-in functionality to fetch nf-core pipelines. You can use the following command to pull the latest version of a remote workflow from the nf-core github site.;
$ nextflow pull nf-core/<PIPELINE>
Nextflow will also automatically fetch the pipeline code when you run
$ nextflow run nf-core/<pipeline>`.
For the best reproducibility, it is good to explicitly reference the pipeline version number that you wish to use with the -revision/-r flag.
In the example below we are pulling the rnaseq pipeline version 3.0
$ nextflow pull nf-core/rnaseq -revision 3.0
We can check the pipeline has been pulled using the nf-core list command.
nf-core list rnaseq -s pulled
We can see from the output we have the latest release.
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 2.1
┏━━━━━━━━━━━━━━━━━━━┳━━━━━━━┳━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━┓
┃ Pipeline Name ┃ Stars ┃ Latest Release ┃ Released ┃ Last Pulled ┃ Have latest release? ┃
┡━━━━━━━━━━━━━━━━━━━╇━━━━━━━╇━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━┩
│ rnaseq │ 394 │ 3.4 │ 4 weeks ago │ 1 minutes ago │ Yes (v3.4) │
[..truncated..]
Development Releases
If not specified, Nextflow will fetch the default git branch. For pipelines with a stable release this the default branch is
master- this branch contains code from the latest release. For pipelines in early development that don’t have any releases, the default branch isdev.
Exercise: Fetch the latest RNA-Seq pipeline
Use the
nextflow pullcommand to download the latestnf-core/rnaseqpipelineUse the
nf-core listcommand to see if you have the latest version of the pipelineSolution
Use the
nextflow pullcommand to download the latestnf-core/rnaseqpipeline$ nextflow pull nf-core/rnaseqUse the
nf-core listcommand to see if you have the latest version of the pipeline$ nf-core list rnaseq --sort pulled
Usage instructions and documentation
You can find general documentation and instructions for Nextflow and nf-core on the nf-core website . Pipeline-specific documentation is bundled with each pipeline in the /docs folder. This can be read either locally, on GitHub, or on the nf-core website.
Each pipeline has its own webpage at https://nf-co.re/
In addition to this documentation, each pipeline comes with basic command line reference. This can be seen by running the pipeline with the parameter --help , for example:
$ nextflow run -r 3.4 nf-core/rnaseq --help
N E X T F L O W ~ version 20.10.0
Launching `nf-core/rnaseq` [silly_miescher] - revision: 964425e3fd [3.4]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/rnaseq v3.0
------------------------------------------------------
Typical pipeline command:
nextflow run nf-core/rnaseq --input samplesheet.csv --genome GRCh37 -profile docker
Input/output options
--input [string] Path to comma-separated file containing information about the samples in the experiment.
--outdir [string] Path to the output directory where the results will be saved.
--public_data_ids [string] File containing SRA/ENA/GEO identifiers one per line in order to download their associated FastQ files.
--email [string] Email address for completion summary.
--multiqc_title [string] MultiQC report title. Printed as page header, used for filename if not otherwise specified.
--skip_sra_fastq_download [boolean] Only download metadata for public data database ids and don't download the FastQ files.
--save_merged_fastq [boolean] Save FastQ files after merging re-sequenced libraries in the results directory.
..truncated..
The nf-core launch command
As can be seen from the output of the help option nf-core pipelines have a number of flags that need to be passed on the command line: some mandatory, some optional.
To make it easier to launch pipelines, these parameters are described in a JSON file, nextflow_schema.json bundled with the pipeline.
The nf-core launch command uses this to build an interactive command-line wizard which walks through the different options with descriptions of each, showing the default value and prompting for values.
Once all prompts have been answered, non-default values are saved to a params.json file which can be supplied to Nextflow using the -params-file option. Optionally, the Nextflow command can be launched there and then.
To use the launch feature, just specify the pipeline name:
$ nf-core launch -r 3.0 rnaseq
Exercise : Create nf-core params file
Use the
nf-core launchcommand to create a params file namednf-params.json.
- Use the
nf-core launchcommand to launch the interactive command-line wizard.- Add an input file name
samples.csv- Add a genome
GRCh38** Note ** : Do not run the command now.Solution
$ nf-core launch rnaseqThe contents of the
nf-params.jsonfile should be{ "input": "samples.csv", "genome": "GRCh38" }
Config files
nf-core pipelines make use of Nextflow’s configuration files to specify how the pipelines runs, define custom parameters and what software management system to use e.g. docker, singularity or conda.
Nextflow can load pipeline configurations from multiple locations. nf-core pipelines load configuration in the following order:

- Pipeline: Default ‘base’ config
- Always loaded. Contains pipeline-specific parameters and “sensible defaults” for things like computational requirements
- Does not specify any method for software packaging. If nothing else is specified, Nextflow will expect all software to be available on the command line.
- Core config profiles
- All nf-core pipelines come with some generic config profiles. The most commonly used ones are for software packaging: docker, singularity and conda
- Other core profiles are debug and two test profiles. There two test profile, a small test profile (nf-core/test-datasets) for quick test and a full test profile which provides the path to full sized data from public repositories.
- Server profiles
- At run time, nf-core pipelines fetch configuration profiles from the configs remote repository. The profiles here are specific to clusters at different institutions.
- Because this is loaded at run time, anyone can add a profile here for their system and it will be immediately available for all nf-core pipelines.
- Local config files given to Nextflow with the
-cflag$ nextflow run nf-core/rnaseq -r 3.0 -c mylocal.config
- Command line configuration: pipeline parameters can be passed on the command line using the
--<parameter>syntax.$ nextflow run nf-core/rnaseq -r 3.0 --email "my@email.com"`
Config Profiles
nf-core makes use of Nextflow configuration profiles to make it easy to apply a group of options on the command line.
Configuration files can contain the definition of one or more profiles. A profile is a set of configuration attributes that can be activated/chosen when launching a pipeline execution by using the -profile command line option. Common profiles are conda, singularity and docker that specify which software manager to use.
Multiple profiles are comma-separated. When there are differing configuration settings provided by different profiles, the right-most profile takes priority.
$ nextflow run nf-core/rnaseq -r 3.0 -profile test,conda
$ nextflow run nf-core/rnaseq -r 3.0 -profile <institutional_config_profile>, test, conda
Note The order in which config profiles are specified matters. Their priority increases from left to right.
Multiple Nextflow configuration locations
Be clever with multiple Nextflow configuration locations. For example, use
-profilefor your cluster configuration, the file$HOME/.nextflow/configfor your personal config such asparams.emailand a working directory >nextflow.configfile for reproducible run-specific configuration.
Exercise create a custom config
Add the
params.emailto a file callednfcore-custom.configSolution
A line similar to one below in the file custom.config
params.email = "myemail@address.com"
Running pipelines with test data
The nf-core config profile test is special profile, which defines a minimal data set and configuration, that runs quickly and tests the workflow from beginning to end. Since the data is minimal, the output is often nonsense. Real world example output are instead linked on the nf-core pipeline web page, where the workflow has been run with a full size data set:
$ nextflow run nf-core/<pipeline_name> -profile test
Software configuration profile
Note that you will typically still need to combine this with a software configuration profile for your system - e.g.
-profile test,conda. Running with the test profile is a great way to confirm that you have Nextflow configured properly for your system before attempting to run with real data
Using nf-core pipelines offline
Many of the techniques and resources described above require an active internet connection at run time - pipeline files, configuration profiles and software containers are all dynamically fetched when the pipeline is launched. This can be a problem for people using secure computing resources that do not have connections to the internet.
To help with this, the nf-core download command automates the fetching of required files for running nf-core pipelines offline.
The command can download a specific release of a pipeline with -r/--release .
By default, the pipeline will download the pipeline code and the institutional nf-core/configs files.
If you specify the flag --singularity, it will also download any singularity image files that are required (this needs Singularity to be installed). All files are saved to a single directory, ready to be transferred to the cluster where the pipeline will be executed.
$ nf-core download nf-core/rnaseq -r 3.4
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 2.1
INFO Saving nf-core/rnaseq download.py:148
Pipeline release: '3.4'
Pull singularity containers: 'No'
Output file: 'nf-core-rnaseq-3.4.tar.gz'
INFO Downloading workflow files from GitHub download.py:151
INFO Downloading centralised configs from GitHub download.py:155
INFO Compressing download.. download.py:166
INFO Command to extract files: tar -xzf nf-core-rnaseq-3.4.tar.gz download.py:653
INFO MD5 checksum for nf-core-rnaseq-3.4.tar.gz: f0e0c239bdb39c613d6a080f1dee88e9
Exercise Run a test nf-core pipeline
Run the nf-core/hlatyping pipeline release 1.2.0 with the provided test data using the profile
testandconda. Add the parameter--max_memory 3Gon the command line. Include the config file,nfcore-custom.config, from the previous exercise using the option-c, to send an email when your pipeline finishes.$ nextflow run nf-core/hlatyping -r 1.2.0 -profile test,conda --max_memory 3G -c nfcore-custom.configThe pipeline does next-generation sequencing-based Human Leukozyte Antigen (HLA) typing and should run quickly.
Solution
$ nextflow run nf-core/hlatyping -r 1.2.0 -profile test,conda --max_memory 3GN E X T F L O W ~ version 21.04.0 Launching `nf-core/hlatyping` [pedantic_engelbart] - revision: 6998794795 [1.2.0] BAM file format detected. Initiate remapping to HLA alleles with yara mapper. ---------------------------------------------------- ,--./,-. ___ __ __ __ ___ /,-._.--~' |\ | |__ __ / ` / \ |__) |__ } { | \| | \__, \__/ | \ |___ \`-._,-`-, `._,._,' nf-core/hlatyping v1.2.0 ---------------------------------------------------- Pipeline Release : 1.2.0 Run Name : pedantic_engelbart File Type : BAM Seq Type : dna Index Location : /home/training/.nextflow/assets/nf-core/hlatyping/data/indices/yara/hla_reference_dna IP Solver : glpk Enumerations : 1 Beta : 0.009 Max Memory : 3G Max CPUs : 2 Max Time : 2d Input : https://github.com/nf-core/test-datasets/raw/hlatyping/bam/example_pe.bam Data Type : Paired-End Output Dir : results Launch Dir : /home/training Working Dir : /home/training/work Script Dir : /home/training/.nextflow/assets/nf-core/hlatyping User : training Max Resources : 3G memory, 2 cpus, 2d time per job Config Profile : conda,test Config Profile Description: Minimal test dataset to check pipeline function Config Files : /home/training/.nextflow/assets/nf-core/hlatyping/nextflow.config, /home/training/nextflow.config, /home/training/.nextflow/assets/nf-> > > >core/hlatyping/nextflow.config ---------------------------------------------------- BAM file format detected. Initiate remapping to HLA alleles with yara mapper. [- ] process > remap_to_hla - executor > local (6) [05/084b41] process > remap_to_hla (1) [100%] 1 of 1 ✔ [5a/9bec8b] process > make_ot_config [100%] 1 of 1 ✔ [54/8bc5d7] process > run_optitype (1) [100%] 1 of 1 ✔ [a9/2cbea8] process > output_documentation [100%] 1 of 1 ✔ [df/d3dac5] process > get_software_versions [100%] 1 of 1 ✔ [e1/903ed9] process > multiqc (1) [100%] 1 of 1 ✔ -[nf-core/hlatyping] Pipeline completed successfully- WARN: To render the execution DAG in the required format it is required to install Graphviz -- See http://www.graphviz.org for more info. Completed at: 26-Oct-2021 10:07:27 Duration : 4m 14s CPU hours : (a few seconds) Succeeded : 6
Troubleshooting
If you run into issues running your pipeline you can you the nf-core website to troubleshoot common mistakes and issues https://nf-co.re/usage/troubleshooting .
Extra resources and getting help
If you still have an issue with running the pipeline then feel free to contact the nf-core community via the Slack channel .
The nf-core Slack organisation has channels dedicated for each pipeline, as well as specific topics (eg. #help, #pipelines, #tools, #configs and much more).
The nf-core Slack can be found at https://nfcore.slack.com (NB: no hyphen in nfcore!). To join you will need an invite, which you can get at https://nf-co.re/join/slack.
You can also get help by opening an issue in the respective pipeline repository on GitHub asking for help.
If you have problems that are directly related to Nextflow and not our pipelines or the nf-core framework tools then check out the Nextflow gitter channel or the google group.
Referencing a Pipeline
Publications
If you use an nf-core pipeline in your work you should cite the main publication for the main nf-core paper, describing the community and framework, as follows:
The nf-core framework for community-curated bioinformatics pipelines. Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen. Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x. ReadCube: Full Access Link
Many of the pipelines have a publication listed on the nf-core website that can be found here.
DOIs
In addition, each release of an nf-core pipeline has a digital object identifiers (DOIs) for easy referencing in literature The DOIs are generated by Zenodo from the pipeline’s github repository.
Key Points
nf-core is a community-led project to develop a set of best-practice pipelines built using the Nextflow workflow management system.
The nf-core tool (
nf-core) is a suite of helper tools that aims to help people run and develop nf-core pipelines.nf-core pipelines can be found using
nf-core list, or by checking the nf-core website.
nf-core launch nf-core/<pipeline>can be used to write a parameter file for an nf-core pipeline. This can be supplied to the pipeline using the-params-fileoption.An nf-core workflow is run using
nextflow run nf-core/<pipeline>syntax.nf-core pipelines can be reconfigured by using custom config files and/or adding command line parameters.
Nextflow coding practices
Overview
Teaching: 30 min
Exercises: 15 minQuestions
How do I make my code readable?
How do I make my code portable?
How do I make my code maintainable?
Objectives
Learn how to use whitespace and comments to improve code readability.
Understand coding pitfalls that reduce portability.
Understand coding pitfalls that reduce maintainability.
Nextflow coding practices
Nextflow is a powerful flexible language that one can code in a variety of ways. This can lead to poor practices in coding. For example, this can lead to the workflow only working under certain configurations or execution platforms. Alternatively, it can make it harder for someone to contribute to a codebase, or for you to amend two years later for article submission. These are some useful coding tips that make maintaining and porting your workflow easier.
Use whitespace to improve readability.
Nextflow is generally not sensitive to whitespace in code. This allows you to use indentation, vertical spacing, new-lines, and increased spacing to improve code readability.
#! /usr/bin/env nextflow
// Tip: Allow spaces around assignments ( = )
nextflow.enable.dsl = 2
// Tip: Separate blocks of code into groups with common purpose
// e.g., parameter blocks, include statements, workflow blocks, process blocks
// Tip: Align assignment operators vertically in a block
params.reads = ''
params.gene_list = ''
params.gene_db = 'ftp://path/to/database'
// Tip: Align braces or instruction parts vertically
include { BAR } from 'modules/bar'
include { TAN as BAZ } from 'modules/tan'
workflow {
// Tip: Indent process calls
// Tip: Use spaces around process/function parameters
FOO ( Channel.fromPath( params.reads, checkIfExists: true ) )
BAR ( FOO.out )
// Tip: Use vertical spacing and indentation for many parameters.
BAZ (
Channel.fromPath( params.gene_list, checkIfExists: true ),
FOO.out,
BAR.out,
file( params.gene_db, checkIfExists: true )
)
}
// Tip: Uppercase process names help readability
process FOO {
// Tip: Separate process parts into distinct blocks
input:
path fastq
output:
path "*.fasta"
script:
prefix = fastq.baseName
"""
tofasta $fastq > $prefix.fasta
"""
}
Improve the workflow readability
Use whitespace to improve the readability of the following code.
#! /usr/bin/env nextflow nextflow.enable.dsl=2 params.reads = '' workflow { foo(Channel.fromPath(params.reads)) bar(foo.out) } process foo { input: path fastq output: path "*.fasta" script: prefix=fastq.baseName """ tofasta $fastq > $prefix.fasta """ } process bar { input: path fasta script: """ fastx_check $fasta """ }Solution
#! /usr/bin/env nextflow nextflow.enable.dsl = 2 params.reads = '' workflow { FOO ( Channel.fromPath( params.reads ) ) BAR ( FOO.out ) } process FOO { input: path fastq output: path "*.fasta" script: prefix = fastq.baseName """ tofasta $fastq > $prefix.fasta """ } process BAR { input: path fasta script: """ fastx_check $fasta """ }
Use comments
Comments are an important tool to improve readability and maintenance. Use them to:
- Annotate data structures expected in a channel.
- Describe higher level functionality.
- Describe presence/absence of (un)expected code.
- Mandatory and optional process inputs.
workflow ALIGN_SEQ {
take:
reads // queue channel; [ sample_id, [ file(read1), file(read2) ] ]
reference // file( "path/to/reference" )
main:
// Quality Check input reads
READ_QC ( reads )
// Align reads to reference
Channel.empty()
.set { aligned_reads_ch }
if( params.aligner == 'hisat2' ){
ALIGN_HISAT2 ( READ_QC.out.reads, reference )
aligned_reads_ch.mix( ALIGN_HISAT2.out.bam )
.set { aligned_reads_ch }
} else if ( params.aligner == 'star' ) {
ALIGN_STAR ( READ_QC.out.reads, reference )
aligned_reads_ch.mix( ALIGN_STAR.out.bam )
.set { aligned_reads_ch }
}
aligned_reads_ch.view()
emit:
bam = aligned_reads_ch // queue channel: [ sample_id, file(bam_file) ]
}
process COUNT_KMERS {
input:
// Mandatory
tuple val(sample), path(reads) // [ 'sample_id', [ read1, read2 ] ]: Reads in which to count kmers
// Optional
path kmer_table // 'path/to/kmer_table': Table of k-mers to count
...
}
Report tool versions
Software packaging is a hard problem, and it can be difficult for a package to report the versions of all the tools it has. It may also be excessive to report the version of everything included in a package, when only a handful of tools are used. This means that it’s up to us to effectively report the versions of the tools we use to aid reproducibility.
process HISAT2_ALIGN {
...
script:
def HISAT2_VERSION = '2.2.0' // Version not available using command-line
"""
hisat2 ... | samtools ...
cat <<-END_VERSIONS > versions.yml
"${task.process}":
hisat2: $HISAT2_VERSION
samtools: \$( samtools --version 2>&1 | sed 's/^.*samtools //; s/Using.*\$//' )
END_VERSIONS
"""
}
Name output channels
Output channels from processes and workflows can be named
using the emit: keyword, which helps readability.
workflow ALIGN_HISAT2 {
...
emit:
alignment = HISAT2_ALIGN.out.bam
}
process HISAT2_ALIGN {
...
output:
tuple val(sample), path("*.bam"), emit: bam
tuple val(sample), path("*.log"), emit: summary
path "versions.yml" , emit: versions
...
}
Use params.parameters in workflow blocks, not in process blocks
The params variables are accessible from anywhere in a workflow.
They can be useful to provide a wide variety of properties and
decision making options. For example, one could use a params.aligner
variable in a workflow to select a particular alignment tool. This
in turn could be coded like:
process ALIGN {
input:
tuple val(sample), path(reads)
path index
...
script:
if ( params.aligner == 'hisat2' ) {
"""
hisat2 ... | samtools ...
...
"""
} else if ( params.aligner == 'star' ){
"""
star ...
...
"""
}
}
A better practice is to use it as an input value.
process ALIGN {
input:
tuple val(sample), path(reads)
path index
val aligner // params.aligner is provided as a third parameter
...
script:
if ( aligner == 'hisat2' ) {
"""
hisat2 ... | samtools ...
...
"""
} else if ( aligner == 'star' ){
"""
star ...
...
"""
}
}
This allows one to see from the workflow block where all
parameters are being used, making the workflow easier to maintain.
There is also a danger that one could modify params variables during
pipeline execution, potentially leading to unreproducible results
in more complex workflows.
All input files/directories should be a process input
Depending on the platform you execute your workflow, files may be easily accessible over the network, or downloadable from the internet. However not all execution platforms support this. The example below could work well on your system, but fail on another (for example compute nodes without internet connection).
process READ_CHECK {
input:
tuple val(sample), path(reads)
...
script:
"""
wget ftp://path/to/database
check_reads $reads /local/copy/database > $sample.report
...
"""
}
A strength of Nextflow is file staging, i.e., preparing files for use in process tasks. Staging files by providing them as process input has several benefits.
- Files are updated only when necessary.
- A single file/folder can be shared, without downloading multiple times as in the example above.
- Nextflow supports retrieving files from any valid URL, meaning potentially fewer lines of code.
process READ_CHECK {
input:
tuple val(sample), path(reads)
path database
...
script:
"""
check_reads $reads $database > $sample.report
...
"""
}
If may be that a file is an optional input depending on other parameters.
In cases when no file should be provided, one can pass an empty list []
instead.
workflow {
COUNT_KMERS ( reads, [] )
}
process COUNT_KMERS {
input:
// Mandatory
tuple val(sample), path(reads) // [ 'sample_id', [ read1, read2 ] ]: Reads in which to count kmers
// Optional
path kmer_table // 'path/to/kmer_table': Table of k-mers to count
...
}
Avoid lots of short running processes
Many execution platforms are inefficient if a workflow
tries to execute many short running processes. It
can take more time to schedule and request resources
for each small instance than bundling the short processes
into a larger process task. Nextflow provides
convenient channel operators, such as buffer, collate,
collect, and collectFile,
that help group together inputs into batches which
can run for longer with a given requested resource.
The short tasks themselves can also be parallelised
inside a process script using the command-line tools
xargs or parallel.
workflow REFINE_DATA {
take:
datapoints
main:
BATCH_TASK ( datapoints.collate(100) )
}
process BATCH_TASK {
input:
val data
script:
"""
printf "%s\\n" $data | \
xargs -P $task.cpus -I {} \
short_task {}
# Alternative:
# parallel --jobs $task.cpus "short_task {1}" :::: $data
"""
}
Include a test profile
A test profile is a configuration profile that specifies
a short running test data set to check the functionality of
the whole pipeline. It can also demonstrate to users of your
workflow the kinds of inputs and outputs to expect. Another
benefit is the possibility of automated testing of your
workflow, ensuring the workflow keeps working as you add
new functionality.
profiles {
test {
params {
reads = 'https://github.com/my_repo/test/test_reads.fastq.gz'
reference = 'https://github.com/my_repo/test/test_reference.fasta.gz'
}
}
}
Write modules that use existing containers
Using containers for software packaging is strongly recommended, as they are intended to operate the same, irrespective of the operating system it runs on. Writing modules which use existing containers reduces maintenance needed for a workflow, and minimises the need to resolve package conflicts. A helpful resource for this is the bioconda channel for the package manager conda, which provides packaging for many bioinformatics tools. In addition to this, Biocontainers builds both Docker and Singularity images for each tool packaged on the bioconda channel. Multi-package containers (known as mulled containers) can also be created following the instructions on the Multi Package Containers repository.
process FASTQC {
container "${ workflow.containerEngine == 'singularity' ?
'https://depot.galaxyproject.org/singularity/fastqc:0.11.9--0' :
'quay.io/biocontainers/fastqc:0.11.9--0' }"
...
}
Building your own container images should be used as a last resort. A preferred option is to write a conda recipe for the tool to be included in the bioconda channel. This makes the tool available via a package manager, and containers are automatically built for the tool.
Use file compression and temporary disk space when possible
Disk space is often a valuable resource on most compute systems.
When possible, work with compressed files.
There are several useful shell commands and operations
that work well with compressed data, such as gunzip -c and zgrep.
Operations such as command substitution ($( command )), or
process substitution (>( command_list ), or <( command_list))
can also help working with compressed data. Lastly,
named pipes can also be used if the above approaches fail.
process COUNT_FASTA {
input:
path fasta // reference.fasta.gz
script:
"""
zgrep -c '^>' $fasta
"""
}
Another good practice is to use local temporary disk space (also
known as scratch space). Often, the workDir is located on a
shared disk space over a network, which slows down processes
that read and write a lot to disk. Using scratch space not only
speeds up disk I/O, but also saves space in the workDir since only
files which match the process output directive are copied back
across for caching. The process directive process.scratch
can be provided with either a boolean or the path to use
for scratch space.
process {
scratch = '/tmp'
}
Use consistent naming conventions
Using consistent naming conventions greatly helps readability. For example using uppercase for process names helps distinguish them from other workflow components like channels or operators. Here are other suggestions one can follow from Nf-core.
Key Points
Nextflow is not sensitive to whitespace. Use it to layout code for readability.
Use comments and whitespace to group chunks of code to describe big picture functionality.
Report tool versions in the scripts.
Name channel outputs using the
emit:keyword.Avoid
params.parameterin a process. Pass all parameters using input channels.Input files should be passed using input channels.
Group short running commands into a larger process.
Include a test profile which runs the workflow on a small test data set.
Write your processes to reuse existing containers/software bundles.
Use compressed files and temporary disk space when possible.
Use consistent naming conventions.